400-029-0925
400-029-0925后颅窝,这个容纳着小脑、脑干和第四脑室的狭小空间,犹如神经系统的 "战略要地"。当起源于室管膜细胞的肿瘤在此生长,其复杂的解剖毗邻与独特的生物学行为,往往给临床诊疗带来多重挑战。作为儿童中枢神经系统最常见的肿瘤之一,后颅窝室管膜瘤(PFB 组)约占所有室管膜瘤的 60%-70%,其精准分型、规范诊断及个体化治疗,始终是神经肿瘤领域的研究热点。我们将从分子病理分型的新进展切入,系统解析该疾病的诊断标准与治疗难点,为临床实践提供科学参考。

一、PFB组后颅窝室管膜瘤分型体系:从组织学走向分子时代的精准分类
(一)组织学分型:WHO 分级体系的临床价值
根据 2021 年 WHO 中枢神经系统肿瘤分类,后颅窝室管膜瘤的组织学分型仍以细胞形态学特征为基础,分为三个级别:
I 级:室管膜下瘤与黏液乳头型室管膜瘤这两种亚型在 PFB 组中较为罕见,前者由形态一致的圆形细胞组成,呈簇状分布于纤维基质中,生长缓慢,常位于第四脑室底部;后者可见特征性的乳头结构,被覆单层黏液分泌细胞,好发于脊髓圆锥,但后颅窝原发者仅占 1%-2%。二者均表现为低增殖活性(Ki-67<5%),手术全切后预后良好,5 年生存率超过 90%。
II 级:经典型室管膜瘤占后颅窝室管膜瘤的 60%-70%,肿瘤细胞呈梭形或圆形,排列成菊形团或乳头状结构,核分裂象少见(<5 个 / 10HPF),边界相对清晰。尽管组织学表现温和,但约 30%-40% 的病例术后会出现局部复发,尤其当存在分子高危特征时(如 1q 染色体获得)。
III 级:间变型室管膜瘤肿瘤细胞具有明显异型性,核分裂象活跃(≥5 个 / 10HPF),可见微血管增生或坏死灶。Ki-67 标记指数通常 > 15%,侵袭性强,易突破软脑膜向蛛网膜下腔播散。该型约占后颅窝室管膜瘤的 20%-30%,即使接受积极治疗,5 年总生存率仍低于 50%。
(二)分子分型:改写临床实践的革命性进展
传统组织学分型的局限性在于无法准确预测预后与指导治疗,而分子生物学技术的进步揭示了后颅窝室管膜瘤的显著异质性。基于基因表达谱和染色体拷贝数变异,目前公认将 PFB 组分为两大分子亚型:
1. PFA 型(后颅窝室管膜瘤 A 型)
- 分子特征:富集 H3K27me3 失活相关突变(如 EP300、CREBBP),染色体 6q、11q 缺失,10q 获得,CIMP(CpG 岛甲基化表型)阳性。该亚型多见于婴幼儿(<3 岁),占后颅窝室管膜瘤的 40%-50%。
- 临床行为:肿瘤常起源于第四脑室侧隐窝,呈浸润性生长,与脑干粘连紧密,手术全切率仅 40%-50%。尽管组织学多为 II 级,但具有更高的复发风险,5 年无进展生存率不足 40%,对传统放化疗敏感性较低。
2. PFB 型(后颅窝室管膜瘤 B 型)
- 分子特征:以 1q 染色体获得(约 80% 病例)为标志性改变,伴有 NOTCH 通路激活,CIMP 阴性,常见于儿童(3-18 岁)及成人,占比 50%-60%。
- 临床行为:肿瘤多起源于第四脑室顶部,边界相对清晰,手术全切率可达 60%-70%。预后显著优于 PFA 型,5 年总生存率约 70%-80%,对放疗反应较好,但 1q 扩增程度与复发风险正相关(拷贝数越高,复发率越高)。
(三)特殊亚型:罕见类型的临床警示
- 复发 / 转移性室管膜瘤:约 15%-20% 的后颅窝室管膜瘤在病程中出现脑脊液播散(M 期),表现为脊髓或幕上多发病灶,分子特征与原发灶一致,PFA 型更易发生远处转移。
- 伴间充质分化的室管膜瘤:罕见,肿瘤内可见软骨、骨或脂肪成分,需与畸胎瘤鉴别,其生物学行为尚不完全明确,但初步研究显示预后与经典型 II 级相似。

二、PFB组后颅窝室管膜瘤诊断标准:多模态评估构建精准诊疗基石
(一)临床表现:警惕后颅窝占位的典型信号
后颅窝室管膜瘤的症状与肿瘤生长部位及速度密切相关,早期常因脑脊液循环受阻出现颅内压增高表现:
- 头痛与呕吐:晨起加重的搏动性头痛,伴随喷射性呕吐,儿童可表现为烦躁不安、拒食,婴儿可见前囟隆起、头围增大。
- 小脑功能障碍:步态不稳、共济失调、眼球震颤,是肿瘤压迫小脑半球或蚓部的特征性表现。
- 脑干受累症状:复视、面瘫、吞咽困难等 cranial nerve 麻痹,提示肿瘤侵犯脑桥或延髓,多见于 PFA 型。
- 急性脑疝风险:当肿瘤阻塞第四脑室出口,可突发意识障碍、呼吸节律异常,需紧急处理。
(二)影像学检查:MRI 主导的定位与定性诊断
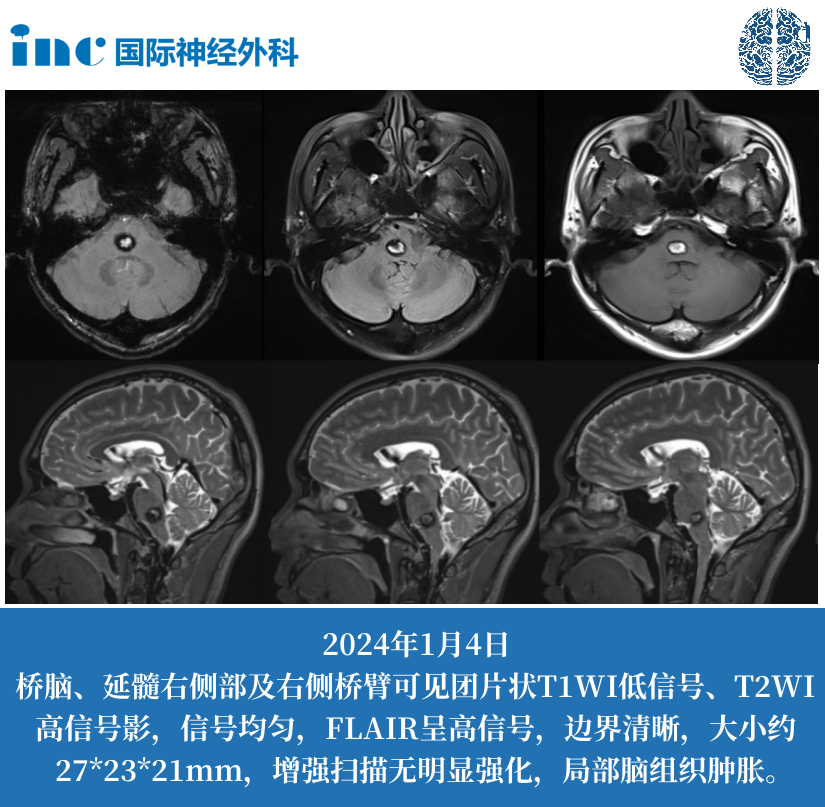
MRI 常规序列
- T1WI:肿瘤多呈等或稍低信号,信号不均匀,可见囊变(20%-30%)、出血(10%-15%)。
- T2WI/FLAIR:呈高信号,瘤周水肿轻(与胶质瘤鉴别点),第四脑室常受压变形,向侧方或后方移位。
- 增强扫描:实性部分呈中度至明显强化,PFA 型强化更不均匀,可见线状或斑片状强化;PFB 型多呈均匀强化,边界较清晰。
特殊序列价值
- DWI(弥散加权成像):鉴别肿瘤实性部分与坏死囊变,室管膜瘤 ADC 值多高于高级别胶质瘤。
- MRS(磁共振波谱):Cho 峰升高,NAA 峰降低,可辅助鉴别诊断,但特异性有限。
鉴别诊断要点
- 髓母细胞瘤:好发于小脑蚓部,儿童多见,T1WI 低信号,增强后明显强化,DWI 呈高信号(细胞密度高)。
- 毛细胞型星形细胞瘤:囊实性为主,壁结节强化,好发于小脑半球,病程较缓慢。
- 血管母细胞瘤:成人多见,富血供肿瘤,可见流空血管影,伴红细胞增多症或 VHL 病史。
(三)病理诊断:从形态学到分子检测的全程质控
术中快速冰冻初步判断肿瘤性质,区分胶质瘤与室管膜瘤,但难以准确分级,需结合术后石蜡切片。
常规病理检查
- 组织形态:经典型可见室管膜菊形团(肿瘤细胞围绕血管排列),间变型可见核分裂象增多、微血管增生。
- 免疫组化:GFAP(+)、S-100(+)、EMA(胞膜点状 +),Ki-67 指数评估增殖活性,≥5% 提示恶性潜能增加。
分子病理检测
- 必做项目:FISH 检测 1q 拷贝数(判断 PFB 型高危因素),DNA 甲基化谱或 RNA 测序区分 PFA/PFB 型(推荐用于儿童患者)。
- 可选项目:检测 MYCN 扩增(罕见,多见于幕上室管膜瘤)、H3K27 突变(提示 PFA 型),为精准分型提供依据。
(四)分期评估:INI 分期与脑脊液播散检测
采用 INI 分期系统(2016 年修订版):
- T 分期:T1(局限于第四脑室),T2(侵犯小脑 / 脑干),T3(突破脑膜)。
- M 分期:M0(无播散),M1(脑脊液细胞学阳性),M2(脊髓转移),M3(颅内转移)。所有患者治疗前需行全脊髓 MRI 及脑脊液细胞学检查(至少 2 次腰椎穿刺),排除播散转移。

三、PFB组后颅窝室管膜瘤治疗难点:从手术困境到系统治疗的多重挑战
(一)手术治疗:在神经保护与肿瘤切除间寻求平衡
1. 解剖学挑战:后颅窝的 "手术雷区"
第四脑室周围毗邻脑干、小脑后下动脉(PICA)、面听神经复合体,肿瘤常沿第四脑室底匍匐生长,尤其是 PFA 型,易侵犯脑桥背侧或延髓,形成 "肿瘤 - 神经血管粘连复合体"。术中过度牵拉可能导致呼吸抑制、面瘫、吞咽障碍等严重并发症,因此全切率受限于解剖边界而非肿瘤生物学行为。
2. 全切率与预后的矛盾关系
尽管国际共识强调 "最大安全切除" 原则,全切率(GTR)仍是最重要的预后因素:PFB 型 GTR 患者 5 年无进展生存率达 70%,而次全切除(STR)者降至 40%;PFA 型即使 GTR,5 年无进展生存率仅 30%-40%。但强行全切可能导致永久性神经功能缺损,尤其对于侵犯脑干的肿瘤,需权衡切除程度与生活质量。
3. 手术技术改进与辅助手段
- 神经电生理监测:术中脑干听觉诱发电位(BAEP)、面神经肌电图(EMG)实时监测,减少颅神经损伤。
- 荧光导航:5-ALA 荧光染色辅助识别肿瘤边界,尤其适用于边界不清的 PFA 型。
- 分阶段手术:巨大肿瘤可先行部分切除缓解脑积水,二期再处理残余病灶,降低单次手术风险。
(二)放射治疗:儿童患者的 "双刃剑" 抉择
1. 术后放疗的适应症争议
PFB 型:无论分级与切除程度,均推荐术后放疗(证据等级 1B),尤其是 1q 扩增患者,放疗可降低 50% 的复发风险。
PFA 型:婴幼儿(<3 岁)放疗获益不明确,因高剂量放疗(≥54Gy)显著增加认知障碍、生长激素缺乏等后遗症,需考虑延迟放疗或化疗替代。
2. 放疗技术与剂量优化
调强放疗(IMRT)/ 质子治疗:精准勾画靶区(瘤床 + 2cm 安全边界),儿童推荐质子治疗以减少颞叶、海马照射剂量,降低远期神经认知损伤。
全脊髓放疗(CSI):仅用于 M 期患者(脑脊液播散),剂量 30-36Gy,原发灶加量至 54-58Gy,需注意脊髓耐受量(儿童≤45Gy)。
3. 放疗并发症管理
急性反应:脑水肿(甘露醇 + 激素)、恶心呕吐(5-HT3 拮抗剂)、脱发(心理支持)。
远期毒性:儿童患者需监测生长发育(每年评估身高、骨龄)、甲状腺功能(每 6 个月查 TSH)、认知功能(每年神经心理测试),30% 的患儿在放疗后 5 年出现不同程度的学习能力下降。
(三)化学治疗:缺乏突破性方案的现状与探索
1. 传统化疗的局限性
辅助化疗:用于婴幼儿(<3 岁)延迟放疗,方案多借鉴婴儿后颅窝室管膜瘤研究,如长春新碱 + 卡铂(VCR+CBP),6-8 周期,客观缓解率约 30%-40%,但无法显著提高 PFA 型生存率。
新辅助化疗:尝试缩小肿瘤体积以提高全切率,证据显示仅对部分 PFB 型有效,对 PFA 型无效。
2. 分子靶向治疗的曙光
1q 扩增抑制剂:针对 PFB 型的 1q 获得,探索 CDK4/6 抑制剂(如 Palbociclib)阻断细胞周期,临床前研究显示可抑制 1q + 肿瘤细胞增殖,II 期临床试验正在进行(NCT03766956)。
EZH2 抑制剂:PFA 型存在 H3K27me3 通路异常,EZH2 抑制剂 Tazemetostat 在动物模型中显示抗肿瘤活性,2023 年 ASCO 报道其单药治疗复发 PFA 型的疾病控制率达 45%。
3. 化疗适应症的精准把控
优先用于:<3 岁患儿(延迟放疗)、PFA 型术后残留、复发 / 转移性病例。
谨慎使用:成人 PFB 型 GTR 患者,因化疗获益不明确,且可能增加第二肿瘤风险(如烷化剂导致白血病)。
(四)复发 / 转移灶的治疗困境
局部复发:首选二次手术,若无法切除则行挽救性放疗(剂量≤50Gy),但脑干受量限制常导致治疗失败。
播散转移:CSI 联合局部加量,有效率仅 20%-30%,新型药物如鞘内注射靶向药物(尚处实验阶段)。
PFA 型复发:缺乏有效治疗手段,中位生存期仅 12-18 个月,需积极推荐入组临床试验。
(五)多学科协作(MDT)的核心价值
后颅窝室管膜瘤的诊疗需神经外科、放疗科、肿瘤内科、儿科、神经心理科等多学科参与:
术前评估:影像科明确肿瘤血供与神经血管关系,病理科快速冰冻指导手术策略。
术后决策:分子病理结果回报后,放疗科制定个体化靶区(如 PFA 型避免过度照射脑干),儿科医生监测儿童生长发育。
长期管理:神经心理科介入放疗后认知干预,康复科制定肢体功能训练方案,提高患者生活质量。
四、PFB组后颅窝室管膜瘤常见疑问
(一)PFA 型与 PFB 型的预后差异为何如此显著?
根本原因在于分子特征的不同:PFB 型的 1q 获得导致致癌基因过表达(如 NOTCH 通路激活),但该通路对放疗敏感;而 PFA 型存在表观遗传调控异常(H3K27me3 丢失),形成 "转录成瘾" 状态,对传统治疗抵抗,且更易发生免疫逃逸。未来需针对 PFA 型的表观遗传靶点开发新药。
(二)儿童患者能否避免放疗?
对于 < 3 岁的 PFB 型患儿,若手术达到 GTR,可观察 3-6 个月,若出现复发迹象再行放疗;PFA 型即使 GTR,复发风险极高,可考虑术后先进行化疗(如 VCR+CBP),至 3 岁后再放疗。近年尝试的免疫治疗(如 PD-1 抑制剂)在儿童中的安全性仍需验证。
(三)如何判断后颅窝室管膜瘤是否发生脑脊液播散?
初次诊断时,需行全脊髓 MRI(包括颈椎、胸椎、腰椎)和 2 次脑脊液细胞学检查(间隔 1 周)。若 MRI 发现脊髓表面结节状强化,或脑脊液找到肿瘤细胞,即可诊断 M 期。需注意,脑脊液标本需采用细胞离心涂片法,提高检出率。
(四)成人后颅窝室管膜瘤的治疗有何特殊之处?
成人病例以 PFB 型为主(约 70%),对放疗反应良好,术后 GTR 者可单纯观察,STR 者推荐放疗(剂量 54-58Gy)。化疗在成人中疗效更差,仅用于复发患者。此外,成人患者需关注放疗对听力的影响(避免耳蜗受量 > 40Gy),术后出现共济失调者可通过康复训练改善平衡功能。
(五)基因检测报告中的 "1q 扩增" 意味着什么?
1q 染色体长臂获得是 PFB 型的标志性改变,约 80% 的 PFB 型患者存在该异常。扩增程度(拷贝数越高)与肿瘤复发风险正相关,且提示对放疗更敏感。因此,1q 状态是制定术后放疗方案的重要依据,尤其是边界切除的患者,需根据 1q 扩增情况决定是否提高照射剂量。
结语
后颅窝室管膜瘤的诊疗历程,见证了从 "组织学粗放分型" 到 "分子精准诊疗" 的跨越式发展。尽管 PFA 型的治疗困境仍待突破,PFB 型的复发风险尚需优化管理,但多学科协作模式与精准医疗技术的结合,正逐步改写患者预后。随着表观遗传药物、免疫治疗的临床转化,以及早期筛查体系的完善,我们有理由相信,后颅窝室管膜瘤将从 "难治性肿瘤" 逐步转化为 "可调控的慢性病"。临床医生需始终保持对分子病理的敏锐洞察,在解剖保护与肿瘤控制间寻找最优平衡,为每一位患者铺就个体化的治疗之路。
- 文章标题:PFB组后颅窝室管膜瘤的分型?诊断标准?治疗难点?
- 更新时间:2025-06-05 18:44:22