400-029-0925
400-029-0925在诊室的柔光下,经常会遇到这样的场景:家长拿着检查报告,目光中满是焦虑与疑惑,"医生,孩子身上的皮疹怎么一直不好,骨头还总说疼,到底得了什么病?" 当病理报告上 "朗格汉斯细胞组织细胞增生症" 的诊断映入眼帘时,这个略显陌生的医学术语背后,究竟隐藏着怎样的疾病密码?作为一种涉及免疫、遗传与环境多重因素的罕见病,朗格汉斯细胞组织细胞增生症(Langerhans Cell Histiocytosis, LCH)的病因探索如同拼图游戏,每一片新发现的拼图都在完善我们对它的认知。本文将从发病机制、临床特征、诊疗策略等多个维度展开,为读者揭开这种疾病的神秘面纱。

一、朗格汉斯细胞组织细胞增生症病因探秘:从细胞异常到系统失衡
(一)基因突变:细胞失控的分子开关
21 世纪初,随着精准医学的兴起,科学家在 LCH 细胞中发现了关键的分子异常 ——BRAF V600E 基因突变。这个位于 7 号染色体的突变位点,如同一个卡住的开关,让丝裂原活化蛋白激酶(MAPK)通路持续激活,导致朗格汉斯细胞异常增殖并逃避凋亡。约 50%-60% 的多系统受累患者和 30% 的单系统病例携带该突变,尤其在儿童患者中更为常见。除了 BRAF,NRAS、MAP2K1 等 MAPK 通路相关基因的突变也逐渐被发现,形成了 "二次打击" 假说:即在遗传易感基础上,获得性体细胞突变打破了细胞增殖的平衡。
这些突变不仅解释了细胞的异常增生,更揭示了 LCH 的克隆性特征。通过激光捕获显微切割技术,研究者在病变组织中检测到单克隆细胞群,证实了这并非单纯的炎症反应,而是具有肿瘤生物学行为的克隆性增殖性疾病。但不同于恶性肿瘤的是,LCH 细胞保留了部分正常朗格汉斯细胞的分化能力,这种 "亦正亦邪" 的特性,正是其临床表现复杂多样的分子基础。
(二)免疫微环境:失控的炎症循环
在显微镜下,LCH 病灶呈现出独特的病理景观:大量异常增生的朗格汉斯细胞与淋巴细胞、浆细胞、嗜酸性粒细胞交织成网,形成肉芽肿性炎症。这种现象提示,免疫系统在疾病发生发展中扮演着双重角色 —— 既是异常增生的诱因,又是自身攻击的对象。
正常情况下,朗格汉斯细胞作为皮肤和黏膜的 "哨兵",负责摄取抗原并激活 T 细胞免疫应答。但在 LCH 患者体内,这种免疫监视机制发生紊乱:Th1 型细胞因子(如 IFN-γ、IL-2)分泌不足,而 Th2 型细胞因子(IL-4、IL-13)异常升高,形成促炎微环境。更关键的是,病变细胞高表达 CD40、CD80 等共刺激分子,持续激活 T 细胞产生 IL-1、TNF-α 等促炎因子,这些细胞因子又反过来促进朗格汉斯细胞增殖,形成恶性循环。这种 "免疫失调 - 细胞增殖 - 炎症放大" 的三角关系,在多系统受累患者中表现得尤为明显,常常导致器官功能损伤。
(三)环境与遗传:迷雾中的潜在诱因
尽管分子生物学研究取得了突破性进展,环境因素的作用仍如同笼罩在病因学上的一层薄雾。流行病学调查显示,LCH 在儿童期发病高峰(1-5 岁)与免疫系统快速发育阶段重合,提示早期环境暴露可能影响疾病易感性。一些研究报道了病毒感染的关联,如人类疱疹病毒 6 型(HHV-6)、EB 病毒在病灶中的检出,但尚未形成明确的因果关系。职业暴露于有机溶剂、重金属的成人患者比例略高,但证据强度仍需更大样本量验证。
遗传易感性研究则聚焦于人类白细胞抗原(HLA)位点和先天性免疫缺陷。家族性 LCH 病例极为罕见,但携带 BRAF 突变的患者亲属中,其他 MAPK 通路相关疾病(如神经纤维瘤病)的发生率略有升高,提示存在遗传易感背景。先天性免疫缺陷病如 GATA2 基因突变综合征患者,LCH 发病风险显著增加,进一步印证了宿主免疫监视功能异常在疾病发生中的基础作用。

二、朗格汉斯细胞组织细胞增生症发病机制:从克隆增殖到器官损伤
(一)细胞起源的争议与厘清
长期以来,朗格汉斯细胞的起源问题存在争议。传统观点认为其属于树突状细胞谱系,起源于骨髓造血干细胞。但最新的单细胞测序研究显示,LCH 细胞具有单核 - 巨噬细胞谱系特征,表达 CD11b、CD14 等髓系标记,这对经典理论提出了挑战。目前学界倾向于认为,LCH 细胞是具有部分树突状细胞表型(如 CD1a、Langerin)的髓系祖细胞异常分化产物,这种分化异常在基因突变和微环境信号的共同作用下发生。
(二)器官受累的病理逻辑
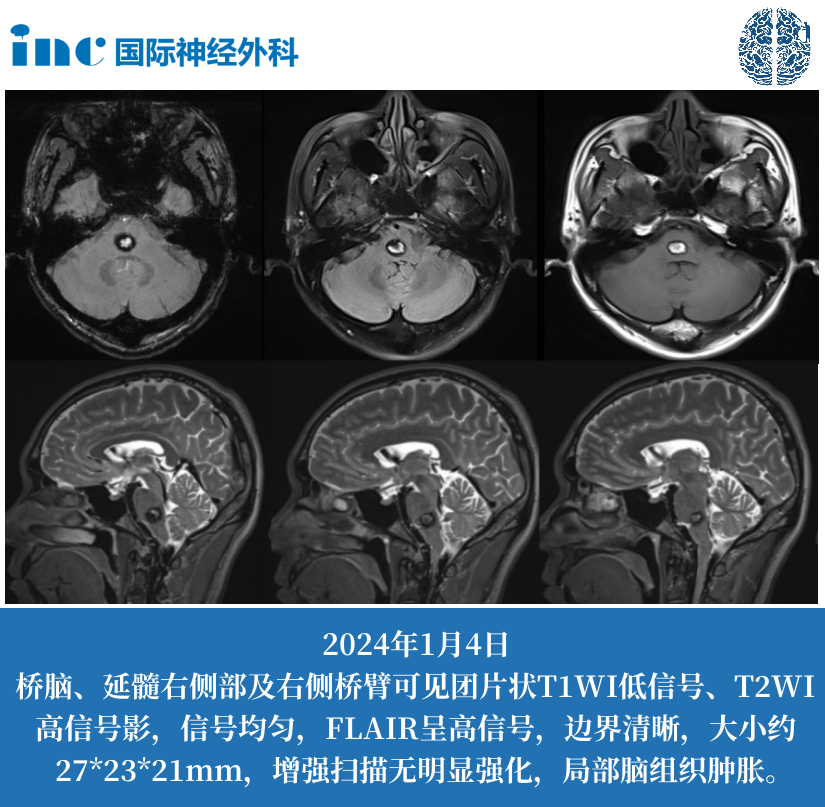
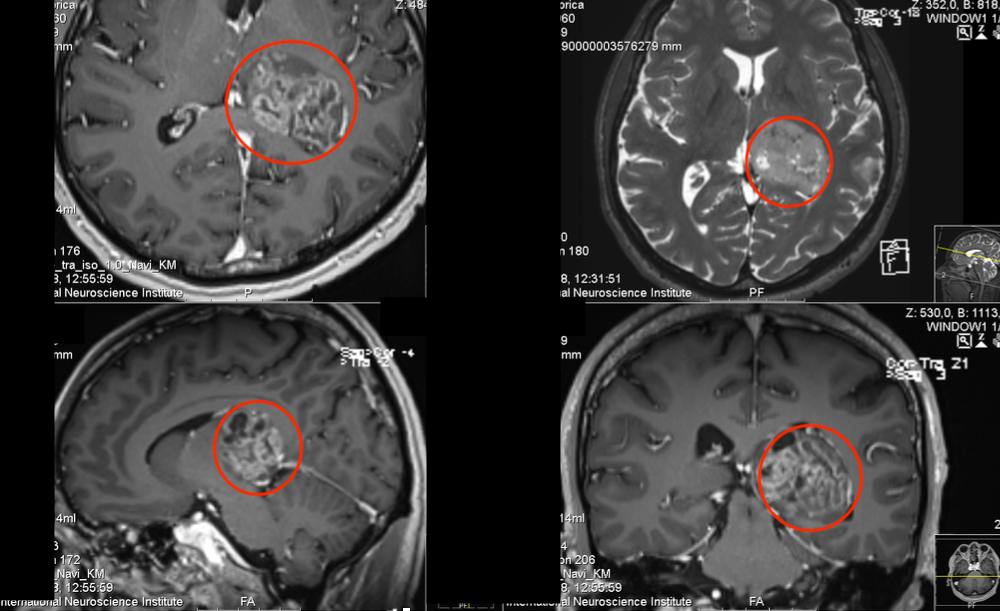
LCH 的临床表现之所以千差万别,与异常细胞的组织嗜性密切相关。骨骼是最常见的受累部位(约 70% 患者),这与破骨细胞活化因子(如 RANKL)的异常分泌有关 ——LCH 细胞通过分泌 RANKL,激活破骨细胞导致溶骨性破坏,形成典型的 "地图样" 骨缺损。在皮肤,异常细胞浸润表皮和真皮浅层,表现为脂溢性皮炎样皮疹或出血性丘疹;在垂体后叶,细胞浸润破坏抗利尿激素分泌,导致烦渴多尿的尿崩症;而在肺部,弥漫性分布的朗格汉斯细胞导致细支气管周围结节和囊腔形成,晚期可发展为肺纤维化。
值得注意的是,不同器官受累的病理进程存在显著差异。单系统病变(如孤立性骨损害)常表现为自限性病程,而多系统受累(尤其是累及造血系统、肝脏、脾脏)则可能进展为危及生命的器官功能衰竭。这种差异不仅与突变类型相关(BRAF 突变更多见于多系统病例),也与微环境中免疫细胞的组成密切相关 —— 例如,肺部病灶中丰富的调节性 T 细胞(Tregs)可能促进了慢性炎症的持续存在。
三、朗格汉斯细胞组织细胞增生症临床症状和系统表现
(一)儿童患者:生长发育中的警示信号
儿童 LCH 占所有病例的 60%-70%,以 1-5 岁幼儿最为常见。最典型的首发症状是骨骼损害,常表现为无痛性肿胀或病理性骨折,颅骨、股骨、肋骨是常见部位。家长可能发现孩子头部出现 "软包",按压时患儿并无明显抗拒,这正是颅骨溶骨性破坏的体征。皮肤表现多样,婴儿期可出现头皮脂溢性皮炎,反复治疗不愈;较大儿童可见躯干四肢的红色丘疹,部分伴有鳞屑或结痂,易被误诊为湿疹或银屑病。
当病变累及下丘脑 - 垂体轴时,约 20%-30% 的患儿会出现尿崩症,表现为饮水次数剧增(每日超过 3L/m² 体表面积)、夜间频繁起尿。眼科受累虽不常见,但眼眶周围骨质破坏可导致突眼,若未及时治疗可能影响视力发育。值得警惕的是多系统受累病例,约 15%-20% 的患儿起病时即表现为发热、体重下降、肝脾肿大,实验室检查可见贫血、血小板减少,这种 "恶性" 表型若延误诊治,死亡率可达 10%-20%。
(二)成人患者:隐匿进展的多面手
成人 LCH 占比约 30%,平均发病年龄 40-50 岁,女性略多于男性。与儿童不同,成人更易累及肺部(约 50%)和淋巴结(30%)。肺部病变早期常表现为干咳、活动后气促,胸部 CT 可见双肺弥漫性小结节和薄壁囊腔,易误诊为肺气肿或肺淋巴管平滑肌瘤病。随着病程进展,囊腔融合导致通气功能障碍,部分患者最终发展为呼吸衰竭。
骨骼损害在成人中多表现为慢性疼痛,腰椎、骨盆等负重部位受累时,疼痛可在活动后加重,夜间休息时缓解,这种与骨关节炎相似的症状常导致诊断延误。皮肤病变相对少见,表现为浸润性斑块或结节,好发于头颈部。值得注意的是,成人多系统受累病例常合并潜在疾病,如血液系统肿瘤(霍奇金淋巴瘤、白血病)或自身免疫病(甲状腺炎、干燥综合征),这种伴随现象提示免疫系统的整体失调可能是重要诱因。
(三)特殊类型:罕见场景下的临床挑战
除了经典型 LCH,临床还可见一些特殊亚型。家族性 LCH 极为罕见,呈常染色体显性或隐性遗传,往往伴有其他遗传性疾病;Letterer-Siwe 病作为急性播散型 LCH,多见于 2 岁以下婴儿,表现为发热、皮疹、肝脾肿大和全血细胞减少,病情凶险;Hand-Schüller-Christian 病则以颅骨缺损、突眼和尿崩症三联征为特征,常见于儿童晚期。这些特殊类型虽占比不足 10%,但临床表现的复杂性对早期诊断提出了更高要求。

四、朗格汉斯细胞组织细胞增生症诊断体系:从形态学观察到分子解码
(一)组织病理:金标准的层层验证
当临床怀疑 LCH 时,组织活检是确立诊断的关键。病理标本在光学显微镜下可见特征性的朗格汉斯细胞:胞质丰富淡染,核形不规则呈肾形或分叶状,核膜有皱褶。但仅凭形态学难以与其他组织细胞疾病鉴别,需通过免疫组化 "三重验证":CD1a 细胞膜阳性、S-100 蛋白胞核胞质阳性、Langerin(CD207)胞质内 Birbeck 颗粒阳性。其中,电镜下观察到球拍状的 Birbeck 颗粒曾被视为确诊依据,但因操作复杂,如今已被免疫组化取代。
(二)影像评估:定位病变的立体地图
影像学检查在评估病变范围和指导活检中发挥重要作用。X 线平片是骨骼病变的初筛手段,典型表现为边界清楚的溶骨性破坏,无硬化边;CT 对肺部微小病变(如 2mm 以下结节)和纵隔淋巴结肿大显示更佳;MRI 则擅长发现软组织受累和垂体柄增粗(尿崩症的特征性表现)。PET-CT 在评估多系统病变活性方面具有优势,高代谢病灶提示疾病处于活动期,可用于疗效监测。
(三)分子检测:精准分型的钥匙
随着靶向治疗的发展,基因检测已成为诊疗流程的重要环节。所有新诊断患者均应检测 BRAF V600E 突变,可通过实时荧光定量 PCR(qPCR)或一代测序实现,突变阳性率约 50%。对于 BRAF 野生型患者,需进一步检测 NRAS、MAP2K1 等其他 MAPK 通路基因,以明确潜在的治疗靶点。值得注意的是,同一患者不同病灶的突变状态可能一致,提示病变起源于单一克隆的播散。
(四)鉴别诊断:排除迷雾中的相似疾病
由于临床表现多样,LCH 需与多种疾病鉴别。在儿童,需排除尤文肉瘤、神经母细胞瘤等恶性肿瘤,以及慢性肉芽肿病等遗传性免疫缺陷;成人则需与肺癌、结节病、淋巴瘤样肉芽肿相鉴别。关键鉴别点在于:LCH 的肉芽肿性炎症以朗格汉斯细胞为主,缺乏恶性细胞的异型性,且免疫组化标记具有特异性。
五、朗格汉斯细胞组织细胞增生症治疗策略:分层管理与精准干预
(一)危险度分层:治疗决策的基石
根据病变范围和器官功能状态,LCH 分为低危、中危和高危组。低危组指单系统单部位受累(如孤立性骨病变),中危组包括单系统多部位或多系统无高危器官(肝、脾、造血系统)受累,高危组则为多系统且伴有上述器官功能损害。这种分层决定了治疗强度:低危患者无需全身治疗,中危患者需局部干预联合轻度全身治疗,高危患者则需强化疗甚至造血干细胞移植。
(二)局部治疗:精准打击局部病灶
对于孤立性骨病变,手术刮除适用于承重部位(如椎体)的小病灶,既能明确诊断又可减轻症状;放疗(剂量 10-20Gy)则用于手术难以切除或复发部位,需注意儿童骨骺部位放疗可能影响生长发育。皮肤病变局部使用糖皮质激素软膏对轻度皮疹有效,结节性病变可考虑病灶内注射糖皮质激素。尿崩症患者需长期使用去氨加压素(弥凝)控制症状,同时监测电解质平衡。
(三)全身治疗:从化疗到靶向的跨越
传统化疗方案:中危患者常用长春碱 + 泼尼松(VP 方案),总有效率约 80%,疗程 6-12 个月。高危患者需更强烈的方案,如 CLAG 方案(克拉屈滨 + 阿糖胞苷)或 DAL-HX 方案,总生存率从过去的 50% 提升至目前的 70%-80%。但化疗的副作用不容忽视,儿童可能出现生长发育迟缓,成人则面临骨髓抑制和肝肾功能损伤。
靶向治疗时代:2014 年,维莫非尼(Vemurafenib)被批准用于 BRAF V600E 突变的难治性 LCH,客观缓解率达 50%-60%,尤其对传统化疗耐药的患者带来希望。达拉非尼(Dabrafenib)联合曲美替尼(Trametinib)针对 BRAF 突变患者的临床试验显示,完全缓解率提升至 30%,且副作用较单药减轻。对于 NRAS 突变患者,MEK 抑制剂司美替尼(Selumetinib)已进入 Ⅲ 期临床,初步数据显示疾病控制率达 75%。
免疫调节治疗:干扰素 -α 曾是二线治疗选择,适用于 BRAF 野生型患者,通过调节 Th1/Th2 平衡抑制细胞增殖,有效率约 30%-40%。近年来,PD-1 抑制剂纳武利尤单抗(Nivolumab)在难治性病例中显示出潜力,其通过阻断 PD-1/PD-L1 通路激活抗肿瘤免疫,尤其对伴有淋巴细胞浸润的病灶效果更佳。
(四)造血干细胞移植:最后的堡垒
对于一线治疗失败、反复复发的高危患者,异基因造血干细胞移植(HSCT)仍是重要选择。欧洲组织细胞协会的研究显示,移植后 5 年无病生存率达 50%-60%,但移植相关死亡率约 15%,主要风险来自感染和器官功能储备不足。预处理方案倾向于降低强度,以减少对儿童生长发育的影响。
(五)长期管理:贯穿生命周期的守护
LCH 患者的随访需持续 5 年以上,尤其是多系统受累者。每年需进行全身体格检查、血液学分析、受累器官功能评估(如肺功能、甲状腺功能),影像学检查根据病变部位每 6-12 个月一次。心理社会支持同样重要,儿童患者需关注生长发育评估和教育融入,成人患者则需指导职业规划和生育咨询(化疗可能影响性腺功能)。
六、朗格汉斯细胞组织细胞增生症预后展望:从生存到生活质量的提升
(一)预后因素的权重分析
单系统 LCH 患者预后良好,5 年生存率超过 95%,多数病例在治疗后 1-2 年内病灶愈合,遗留轻度后遗症(如骨缺损修复后的形态异常)。多系统无高危器官受累者,经过规范治疗,80% 可获得长期缓解,但约 20% 会出现尿崩症等永久性后遗症。高危患者的预后差异较大,早期识别(诊断时存在贫血、血小板减少)和及时启动强化疗可显著改善结局,现代治疗下 5 年生存率已从 1970 年代的 30% 提升至 75%。
(二)未解之谜与研究方向
尽管诊疗水平显著进步,仍有诸多问题亟待解决:为何约 30% 的 BRAF 突变患者对靶向治疗耐药?微环境中的成纤维细胞和血管内皮细胞如何参与疾病进展?表观遗传学调控(如 DNA 甲基化异常)在 LCH 发生中的作用机制?这些问题的答案,将为开发更精准的治疗方案提供线索。目前,全球多中心协作的 LCH-3 研究正在评估新型靶向药物的长期疗效,而单细胞测序和类器官模型的应用,正推动我们从细胞层面理解疾病异质性。
在诊室的灯光下,当我们向患者解释 LCH 的诊疗方案时,手中的不再是冰冷的医学指南,而是基于分子特征和个体差异的精准治疗蓝图。从最初的病理困惑到如今的靶向治疗,人类对这种疾病的认知每前进一步,都意味着更多患者离治愈更近一步。尽管前路仍有未知,但在基础研究与临床实践的双轮驱动下,我们有理由相信,朗格汉斯细胞组织细胞增生症终将从 "罕见难治" 走向 "可防可控",让每个患者都能在精准医疗的阳光下,重获生命的正常节律。
- 文章标题:朗格汉斯细胞组织细胞增生症什么病因?能治好吗?
- 更新时间:2025-06-06 18:52:14