400-029-0925
400-029-0925胚胎肿瘤是一种相对未分化的高度恶性肿瘤,传统上被认为是WHO IV级。虽然历史上被认为是原始神经外胚层肿瘤(PNET),但胚胎肿瘤的范围很广,在临床和生物学上都是独特的实体。因此,cns -胚胎肿瘤指的是髓母细胞瘤、非典型/畸胎样横纹肌瘤(ATRT)、CNS-PNET、伴有多层玫瑰花结的胚胎肿瘤(ETMR)和松果体细胞瘤。本文将主要介绍髓母细胞瘤的综合基因组学。

髓母细胞瘤
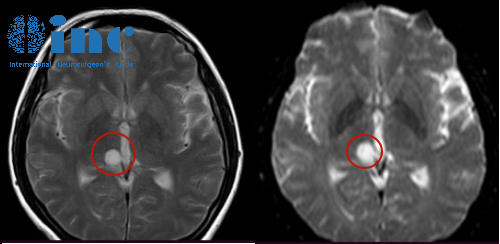
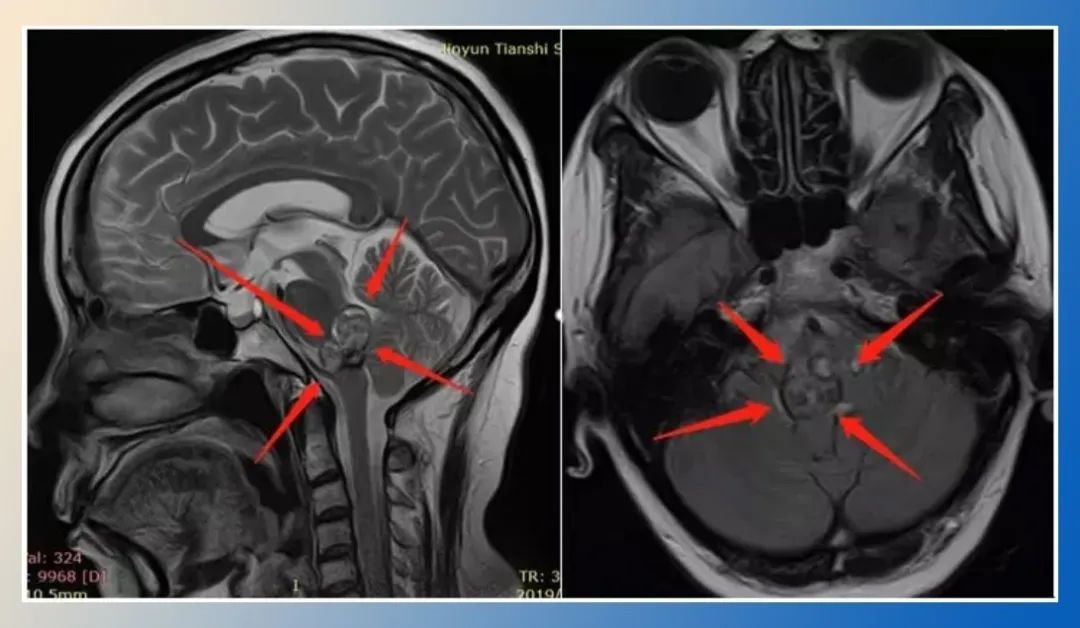
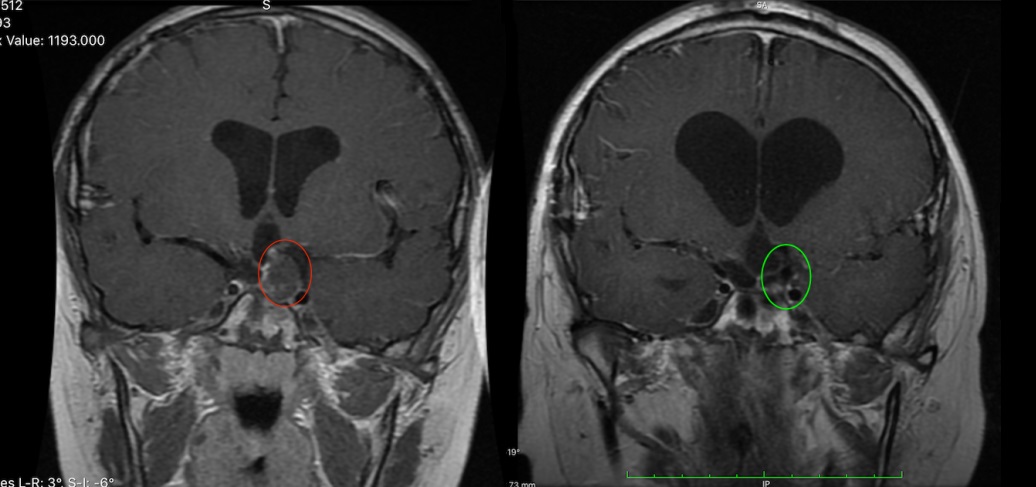
髓母细胞瘤是一种小而圆的蓝色细胞肿瘤,起源于小脑,以前被称为小脑PNET。历史上,髓母细胞瘤被认为是一种形态学实体;最近的研究表明,肿瘤间存在着较大的异质性。存在一种组织学分类,将髓母细胞瘤分为四种组织学亚型,分别为组织增生型、广泛结节性髓母细胞瘤、典型型和大细胞/间变型。然而,组织学分类有几个缺点,特别是缺乏观察者间的可靠性,除了婴儿的纤维组织增生和大龄儿童的程度较轻的纤维组织增生外,缺乏预后的实用价值,并且观察到在儿童中80%的髓母细胞瘤是典型的组织学。临床髓母细胞瘤通常被视为一个实体,3岁以上的患者如果在手术后没有残留疾病或转移灶,则被视为平均/标准风险,否则他们接受更高剂量的颅脊髓照射的风险较低。该方案导致平均/标准风险患者的5年无进展生存率为85%,高危患者的5年无进展生存率为60 - 70%。然而,尽管进行了的治疗,许多患者仍会复发,那些存活下来的患者会留下毁灭性的神经认知和神经内分泌后遗症,是年幼的儿童。
综合基因组学的应用使我们观察到:髓母细胞瘤实际上包括四个不同的亚组,具有不同的转录组、遗传学、人口统计学和结果。这四组被称为WNT(激活无翼/ WNT通路)、SHH(激活Sonic Hedgehog通路)、3组和4组,现在被明确地视为实体,修订后的2016年WHO中枢神经系统肿瘤分类反映了这一新的范式;这些组的识别改变了我们对预后、治疗和临床前建模的方法。
WNT组的特征是WNT通路的激活,大约90%的肿瘤在CTNNB1(编码-连环蛋白的基因)的外显子3中存在激活突变。在发育过程中,它们的细胞来源可能是脑干中的blbp阳性细胞,放射学上它们似乎来自侧隐窝。这种突变被认为是诊断WNT肿瘤的金标准。总的来说,WNT肿瘤有一个相对温和的基因组,其他并发的基因组事件包括monosomy 6、DDX3X突变和TP53突变。WNT肿瘤约占全部髓母细胞瘤的10 - 15%,在世界各地的多个队列中临床表现为预后良好。WNT肿瘤很少转移。通过手术、放疗和基于顺铂的化疗,WNT肿瘤的5年生存率接近全切 。虽然在WNT和SHH肿瘤中都发现了TP53突变,但它们在WNT患者中并不具有预后。Turcot综合征2型定义为WNT激活APC的种系突变,在WNT成神经管母细胞瘤中占很小的比例,WNT肿瘤可以通过免疫组化、CTNNB13外显子测序、单倍体6、全基因组甲基化或转录谱分析等多种方式识别,包括-连环蛋白核聚集。核-catenin和monosomy 6不应该被单独用于诊断WNT肿瘤,因为两者都可以在非WNT肿瘤中被识别。一些临床试验正在进行中,试图通过减少颅脊髓放射剂量来降低WNT肿瘤的治疗。
SHH肿瘤具有SHH通路的激活,起源于颗粒层外,并在小脑内部被发现。SHH肿瘤是婴儿和成人的主要亚组,但在各个年龄段均有发生。下一代测序表明,SHH通路中经常出现突变和拷贝数畸变,包括PTCH1、SMO和SUFU的突变和GLI2的扩增。其他广泛的拷贝数变体包括损失17p, 10q和9q。MYCN扩增也在SHH肿瘤中发现,特别是在儿童中,预示着不良预后。临床上,SHH肿瘤的婴儿组织学为粘结膜增生性,在保留放射治疗的情况下预后良好。患有SHH肿瘤的儿童,如果TP53(体细胞或种系)发生突变,并与GLI2和MYCN扩增同时发生,构成高危的群体,则预后差。成人SHH肿瘤的研究较少,但占成人髓母细胞瘤的80%。SHH通路控制剂,特别是平滑控制剂,正在进行临床试验,似乎对ptch1突变患者等上游事件具有活性;然而,TP53突变的高危患者不太可能因下游事件(如GLI2扩增)而产生反应。SHH肿瘤较常在手术肿瘤床局部复发。
一些家族综合征与SHH髓母细胞瘤相关,包括Gorlin综合征、Li-Fraumeni综合征和种系SUFU突变。全部SHH髓母细胞瘤患者都应紧急进行遗传咨询,以筛选生殖系PTCH1 (Gorlin综合征)、生殖系TP53 (Li-Fraumeni综合征)和生殖系SUFU突变。fanconi相关基因(PALB2, BRCA1/2)的种系事件也可以在一小部分SHH患者中发现。
3组肿瘤多见于婴幼儿,属于预后不良组。它们经常转移,具有大细胞/间变性形态,20%具有MYC癌基因的高水平扩增。在3组肿瘤中缺乏体细胞核苷酸变异,似乎是一个拷贝数驱动的亚组。其他常见的拷贝数畸变包括OTX2和ACVR1/2的扩增,以及广泛的畸变包括同染色体17q和染色体1和9上激活GFI1/1b致癌基因家族的局灶缺失。3组肿瘤复发几乎都是转移性的,没有任何局部手术床复发,特别是在以前接受过放疗的患者中。患有三组肿瘤的婴儿预后特别差,是转移性肿瘤。组3肿瘤可以通过全基因组甲基化或表达和有限的基因表达面板诊断。3组免疫组化不可靠,应避免。
4组是较常见的子组,占3至17岁儿童的40%以上。他们经常转移,几乎80%的4组有17q同染色体。其他细胞遗传学异常包括MYCN和CDK6扩增、7q增加、8p损失和SNCAIP串联复制。与SHH肿瘤相比,4组中MYCN扩增并不预示预后。尽管在某些系列中,平均风险4组肿瘤的患者预后良好,但预后是可变的。11号染色体完整丢失的非转移性4组肿瘤被认为是低风险,11号染色体完整的肿瘤被认为是标准风险,转移性4组被认为是。放疗组4患者仅复发并伴有转移性播散,经常无手术床复发,婴儿组4患者仅接受化疗预后较差,且局部复发。晚期局部复发的4组患者应提示怀疑为辐射诱导的高级别胶质瘤,需要活检。
相关参考资料来源:https://doi.org/10.1007/978-3-319-31512-6_80-1
- 文章标题:髓母细胞瘤的综合基因组学
- 更新时间:2021-06-02 09:54:41