400-029-0925
400-029-0925在人体脑部这个精密的"指挥中心"里,苍白球作为基底神经节的重要组成部分,虽体积不大却肩负着调控运动、维持姿势平衡的关键使命。当这个深藏于大脑深部的结构发生病变时,轻则影响肢体协调,重则导致严重的神经功能障碍,尤其对发育中的儿童而言,病变的影响可能更为深远。
脑部苍白球的病变有哪些?——多维度解析病变类型与病理特征
一、遗传性苍白球病变:基因缺陷引发的神经退行性改变
1.肝豆状核变性(Wilson病)
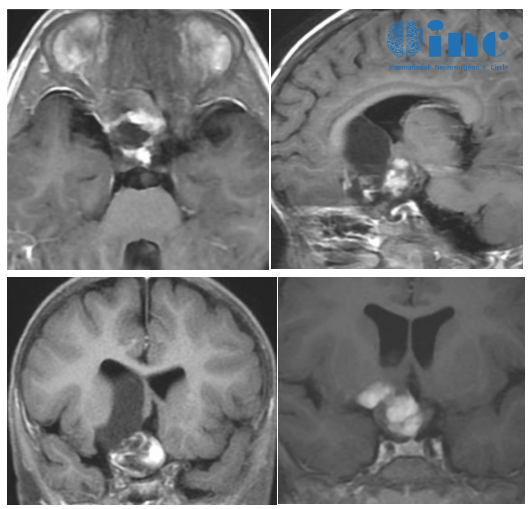
作为一种常染色体隐性遗传的铜代谢障碍性疾病,Wilson病的病理基础在于ATP7B基因变异导致肝细胞内铜转运障碍,过量铜离子在肝、脑等器官沉积。其中苍白球是脑部受累的主要区域之一,铜沉积可引发神经元变性、胶质细胞增生及小血管病变。临床研究显示,约60%的Wilson病患者会出现锥体外系症状,表现为肢体震颤、肌张力增高、动作迟缓等,部分患者可伴有构音障碍及吞咽困难。头颅MRI检查常可见双侧苍白球对称性长T1、长T2信号,此特征性影像学表现有助于疾病诊断。据《中华神经科杂志》2023年发表的流行病学调查数据,我国Wilson病的群体发病率约为1/30000,基因携带率约1/90。
2.泛酸激酶相关神经变性病(PKAN)
这是一种由PANK2基因突变导致的罕见神经退行性疾病,属于neurodegeneration with brain iron accumulation(NBIA)疾病谱的典型代表。异常基因表达使泛酸激酶活性降低,导致铁在苍白球等基底节区异常沉积,形成特征性的"虎眼征"(MRI检查中苍白球内侧低信号伴外侧高信号)。患儿多在青少年时期起病,临床表现为进行性肌张力障碍、舞蹈样动作、帕金森样症状及认知功能减退。美国国立卫生研究院(NIH)的统计显示,PKAN在NBIA疾病中占比约30%-40%,全球发病率约为1/100万。
3.家族性原发性肌张力障碍
部分家族性肌张力障碍类型与苍白球病变相关,如DYT1型肌张力障碍,由TORS2A基因变异引起,病理改变主要为苍白球内侧部(GPi)神经元功能异常。患者多在儿童或青少年期发病,表现为局限性或全身性肌张力障碍,动作时症状加重,休息时减轻。研究表明,DYT1型肌张力障碍在Ashkenazi犹太人群中携带率较高,约为1/300,而在其他人群中发病率约为1/10万。

二、获得性苍白球病变:多种病因导致的结构性与功能性损伤
1.缺血缺氧性脑病(HIE)
围产期窒息、心脏骤停等原因引起的脑缺氧缺血,可导致苍白球选择性损伤。这是由于苍白球对缺氧缺血的敏感性较高,其血供主要来自豆纹动脉的深穿支,该血管在缺氧时易发生痉挛或闭塞。新生儿HIE患者中,约40%-60%会出现苍白球受累,临床表现为肌张力异常、惊厥、意识障碍等。影像学检查可见双侧苍白球对称性异常信号,严重者可出现囊变坏死。《Neurology》杂志2022年的一项研究显示,成年患者因心脏骤停导致的HIE中,苍白球损伤发生率达35%,且与预后不良显著相关。
2.中毒性病变
某些毒素可特异性损伤苍白球,常见类型包括:
一氧化碳中毒:CO与血红蛋白结合形成碳氧血红蛋白,导致组织缺氧,同时CO可抑制细胞色素氧化酶活性,直接损伤神经元。苍白球对缺氧尤为敏感,约20%-30%的重度CO中毒患者会出现双侧苍白球对称性坏死,临床表现为迟发性脑病,即在急性中毒恢复后2-60天出现认知障碍、帕金森综合征等。
锰中毒:长期接触锰粉尘的职业人群(如矿工、电焊工)易发生慢性锰中毒,锰离子可通过血脑屏障在苍白球等基底节区蓄积,导致神经元变性。临床早期表现为注意力不集中、记忆力减退,后期可出现肌张力增高、动作笨拙等帕金森样症状。头颅MRI可见苍白球T1加权像高信号,此为锰沉积的特征性表现。世界卫生组织(WHO)建议工作环境中锰的时间加权平均浓度应低于0.1mg/m³,以降低中毒风险。
3.感染与炎症性病变
病毒性脑炎:某些病毒(如乙型脑炎病毒、单纯疱疹病毒)感染可累及苍白球,引发神经元炎症反应及坏死。乙型脑炎患者中,约15%-20%会出现基底节受累,临床表现为肢体震颤、肌张力增高。
自身免疫性疾病:如抗N-甲基-D-天冬氨酸受体(NMDAR)脑炎,少数患者可伴有苍白球异常信号,可能与自身抗体介导的神经炎症有关。
4.代谢性疾病
糖尿病酮症酸中毒:严重高血糖及酮症状态可导致代谢紊乱,部分患者可出现苍白球对称性损伤,机制可能与缺血、酸中毒及能量代谢障碍有关。临床表现为急性起病的肌张力异常、舞蹈样动作,血糖控制后症状可部分缓解。
线粒体脑肌病:如Leigh综合征,是一种由线粒体DNA或核基因缺陷引起的代谢性疾病,苍白球、丘脑等部位易受累,病理表现为神经元变性及海绵状改变。患儿多在1-2岁起病,表现为发育倒退、肌张力低下、呼吸异常等,预后较差。
三、血管性苍白球病变:脑血管异常引发的急性与慢性损伤
1.脑出血与脑梗死
苍白球的血供来自豆纹动脉的深穿支,这些细小血管易因高血压、动脉粥样硬化等原因发生破裂或闭塞。苍白球区脑出血约占高血压性脑出血的5%-10%,患者多突然出现对侧肢体偏瘫、偏身感觉障碍,若累及内囊后肢可伴有偏盲,即"三偏综合征"。而苍白球区脑梗死相对少见,临床表现与出血类似,但症状通常较轻,进展较缓慢。《中国脑血管病杂志》2021年数据显示,基底节区卒中占所有缺血性卒中的25%-30%,其中苍白球受累约占15%。
2.海绵状血管瘤
这是一种由异常血管团组成的血管畸形,可发生于苍白球等脑内任何部位。患者多因反复少量出血或癫痫发作就诊,头颅MRI表现为边界清楚的混杂信号影,周围可见含铁血黄素沉积环。苍白球海绵状血管瘤的手术切除难度较大,需考虑邻近重要结构(如内囊)的保护。
四、肿瘤性苍白球病变:原发性与转移性肿瘤的浸润
1.原发性脑肿瘤
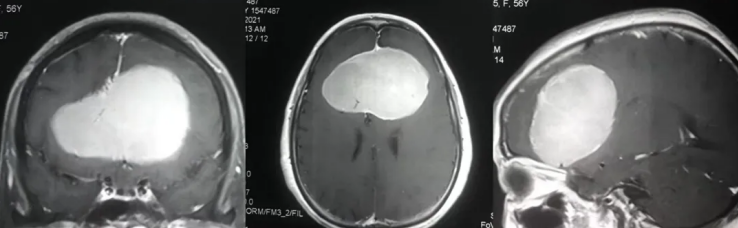
胶质瘤:苍白球原发性胶质瘤相对少见,多为低级别星形细胞瘤或少突胶质细胞瘤,临床表现为缓慢进展的运动障碍、认知功能下降。手术切除是主要治疗手段,但因位置深在,全切难度较大,术后常需辅助放疗或化疗。
神经节细胞瘤:为良性肿瘤,由成熟神经元组成,生长缓慢,可发生于苍白球,多表现为癫痫发作或局灶性神经功能障碍。
2.转移性肿瘤
全身其他部位的恶性肿瘤(如肺癌、乳腺癌)可通过血行转移至苍白球,形成转移瘤。患者多有原发肿瘤病史,临床表现为急性起病的神经功能缺损,头颅MRI可见单发或多发结节,周围伴有明显水肿。
小孩苍白球病变能改善吗?——儿童患者的干预策略与预后分析
一、儿童苍白球病变的特点与诊断要点
1.发病机制的特殊性
与成人相比,儿童苍白球病变的病因更多与遗传性、先天性因素相关,如Wilson病、PKAN、Leigh综合征等,而获得性因素(如缺血缺氧、中毒)在新生儿及婴幼儿期更为常见。首都医科大学附属北京儿童医院的临床数据显示,在该院收治的苍白球病变患儿中,遗传性疾病占比达65%,其中Wilson病和PKAN最为常见,而新生儿HIE导致的苍白球损伤占获得性病变的70%。
2.临床表现的多样性
儿童苍白球病变的症状表现与年龄密切相关:
新生儿期:主要表现为肌张力异常(增高或降低)、喂养困难、惊厥发作,严重者可出现昏迷。
婴幼儿期:可出现运动发育迟缓,如坐、站、走等里程碑延迟,同时伴有姿势异常(如角弓反张)、不自主运动(如震颤、舞蹈样动作)。
学龄前期及学龄期:更多表现为肌张力障碍、动作笨拙、书写困难,部分患儿可伴有认知功能损害及行为异常。
3.诊断流程的规范化
儿童苍白球病变的诊断需遵循"临床-影像-基因-代谢"的多维度评估:
1.详细采集病史:包括围产期情况、家族史、发育史及症状进展特点。
2.神经系统查体:重点评估肌张力、姿势反射、不自主运动及认知功能。
3.影像学检查:头颅MRI是首选检查,可显示苍白球的信号改变、形态异常及受累范围,必要时可行MRS(磁共振波谱分析)评估代谢变化。
4.实验室检查:包括血尿常规、肝肾功能、血铜、尿铜(Wilson病筛查)、血清乳酸(线粒体疾病筛查)等。
5.基因检测:对遗传性病变具有确诊价值,如PKAN需检测PANK2基因,DYT1型肌张力障碍需检测TORS2A基因。
6.代谢筛查:必要时行尿有机酸分析、血氨基酸谱分析等,排除代谢性疾病。
二、儿童苍白球病变的治疗策略:多学科协作的个体化干预
1.病因治疗:针对不同病因的特异性干预
遗传性疾病:
Wilson病:早期诊断后需立即启动驱铜治疗,常用药物包括青霉胺、曲恩汀,同时限制高铜饮食(如动物肝脏、坚果等)。研究显示,早期治疗可使80%以上的患儿病情得到控制,神经症状改善率达60%。《中华儿科杂志》2022年指南建议,Wilson病患儿需终身治疗,并定期监测血铜、肝功能及24小时尿铜。
PKAN:目前尚无特效治疗药物,主要为对症支持治疗,如使用多巴类药物改善帕金森样症状,应用肉毒毒素缓解肌张力障碍。近年来,基因治疗在动物模型中显示出一定前景,但临床应用仍处于研究阶段。
获得性疾病:
HIE:新生儿期需积极改善脑缺氧缺血状态,包括亚低温治疗(适用于中重度HIE)、神经营养支持等。早期干预(如出生后24小时内启动亚低温治疗)可使患儿神经发育预后改善率提高30%-40%。
中毒性病变:如CO中毒,需立即脱离中毒环境,给予高压氧治疗,同时应用神经营养药物。研究表明,早期高压氧治疗可减少迟发性脑病的发生风险,改善率达50%-60%。
2.对症治疗:缓解症状与改善功能
运动障碍的管理:
药物治疗:对于肌张力增高者,可使用巴氯芬、苯海拉明等;对于震颤明显者,可应用普萘洛尔;对于舞蹈样动作,可选用氟哌啶醇。
肉毒毒素注射:适用于局限性肌张力障碍,如痉挛性斜颈、手指肌张力障碍,可缓解肌肉痉挛,改善动作功能。
康复训练:是儿童苍白球病变治疗的重要组成部分,包括物理治疗(PT)、作业治疗(OT)、言语治疗(ST)等。北京博爱医院的临床数据显示,系统康复训练可使70%的患儿运动功能得到不同程度改善,其中5岁以下患儿的康复效果更为显著。
认知与行为干预:对于伴有认知功能损害及行为异常的患儿,需联合心理科医生进行认知行为治疗(CBT),必要时可使用改善认知的药物(如哌甲酯)。
3.手术治疗:精准干预的进阶选择
脑深部电刺激术(DBS):对于药物治疗效果不佳的严重肌张力障碍患儿,DBS可作为备选方案。靶点通常选择苍白球内侧部(GPi)或丘脑底核(STN),通过电刺激调节异常的神经环路。临床研究显示,DBS可使60%-70%的患儿肌张力障碍症状得到显著改善,且儿童患者的疗效优于成人。
立体定向毁损术:如苍白球毁损术(PVP),因可能产生不可逆损伤及较多并发症,目前已较少应用于儿童患者。
4.支持治疗:改善生活质量的基础保障
营养支持:根据患儿具体情况制定个性化饮食方案,如Wilson病需低铜饮食,线粒体疾病需高能量、低脂肪饮食。
家庭护理:指导家长进行正确的体位摆放、关节活动度训练及日常生活护理,预防压疮、关节挛缩等并发症。
教育支持:为患儿提供特殊教育服务,帮助其融入学校生活,最大限度发挥潜能。
三、儿童苍白球病变的预后影响因素与长期管理
1.预后相关因素分析
病因类型:遗传性疾病中,Wilson病若早期治疗预后较好,5年生存率可达90%以上;而PKAN、Leigh综合征等预后较差,多数患儿在数年内病情进展,甚至危及生命。获得性病变中,HIE的预后与缺氧程度及干预时机密切相关,轻度HIE患儿多数可正常发育,中重度者约30%-50%会遗留不同程度的神经功能障碍。
发病年龄:发病年龄越小,对脑发育的影响越大,预后相对较差。新生儿期发病的患儿神经发育迟滞的发生率达80%,而学龄期发病者仅为30%。
干预时机与方法:早期诊断、规范治疗是改善预后的关键。如Wilson病在症状出现前即开始治疗,可完全避免神经损伤;HIE患儿在出生后6小时内启动亚低温治疗,可使死亡率降低40%。
2.长期随访与管理要点
定期评估:患儿需定期(每3-6个月)进行神经系统查体、发育评估及影像学复查,监测病情进展及治疗效果。
治疗方案调整:根据病情变化及时调整药物剂量及康复方案,如随着年龄增长,患儿的肌张力障碍可能从局限性发展为全身性,需相应调整肉毒毒素注射部位及剂量。
并发症防治:长期随访中需关注并发症的发生,如Wilson病患儿可能出现肝功能损害、溶血性贫血,需定期监测肝功能及血常规;DBS术后患儿需注意电极移位、感染等并发症。
心理支持:随着年龄增长,患儿可能因肢体功能障碍、认知缺陷产生心理问题,需及时进行心理干预,必要时联合精神科医生治疗。
四、儿童苍白球病变的康复案例与前沿进展
前沿治疗技术进展
干细胞治疗:间充质干细胞(MSC)具有免疫调节、神经再生等功能,在动物实验中已显示出对苍白球损伤的修复作用。2023年《Stem Cell Research&Therapy》发表的一项临床研究显示,静脉输注MSC可改善HIE患儿的神经发育预后,且安全性良好。
基因编辑技术:如CRISPR-Cas9系统,在PKAN等遗传性苍白球病变的动物模型中,已成功修复突变基因,减少铁沉积,改善神经功能。虽然临床转化仍面临诸多挑战,但为根治遗传性病变带来了希望。
虚拟现实(VR)康复:将VR技术应用于儿童康复训练,通过沉浸式体验提高患儿的训练积极性,改善动作协调性及认知功能。研究显示,VR康复可使患儿的运动功能改善率提高15%-20%。
什么的病变部位在苍白球?——解析累及苍白球的疾病谱与病理机制
一、遗传性神经退行性疾病:基因异常导致的苍白球特异性损伤
1.神经铁沉积性neurodegeneration(NBIA)
除前文提及的PKAN外,NBIA疾病谱还包括:
泛酸激酶相关神经变性病2型(PKAN2):由PLA2G6基因突变引起,临床表现与PKAN类似,但发病年龄较晚,影像学上苍白球铁沉积不如PKAN明显。
脂肪酸羟化酶相关神经变性病(FAHN):由FA2H基因突变导致,苍白球、黑质等部位受累,患者多在儿童期起病,表现为肌张力障碍、帕金森样症状及认知衰退。
Kufor-Rakeb综合征(KRS):为ATP13A2基因突变所致,除苍白球铁沉积外,还伴有黑质病变,临床表现为帕金森综合征、痴呆及锥体束征。
2.亨廷顿病(Huntington disease,HD)
这是一种由HTT基因CAG重复序列异常扩展引起的常染色体显性遗传病,病理改变主要为纹状体(尤其是尾状核和壳核)神经元变性,但随着病情进展,苍白球也会受到累及。患者多在中年起病,但约5%-10%的病例为青少年型HD,表现为迅速进展的肌张力障碍、癫痫发作及认知衰退。苍白球受累可能与纹状体-苍白球环路功能异常有关,导致运动调控失衡。
3.共济失调-毛细血管扩张症(Ataxia-telangiectasia,AT)
由ATM基因突变引起,是一种罕见的常染色体隐性遗传病,主要影响神经系统、免疫系统及血管系统。苍白球、小脑等部位可出现神经元变性及胶质增生,患者多在儿童期起病,表现为进行性共济失调、眼球运动障碍,部分患儿可伴有舞蹈样动作及肌张力障碍。
二、代谢性疾病:能量代谢紊乱引发的苍白球继发性损伤
1.线粒体脑肌病
除Leigh综合征外,其他类型的线粒体脑肌病如MELAS(线粒体肌病伴乳酸血症和卒中样发作)、MERRF(肌阵挛性癫痫伴破碎红纤维)等,在疾病晚期也可累及苍白球。线粒体功能异常导致能量生成障碍,苍白球等对能量需求较高的区域易受影响,出现神经元变性及髓鞘脱失。临床表现除肌肉无力、癫痫发作外,还可出现运动障碍及认知功能下降。
2.戊二酸血症I型(Glutaric acidemia type I)
因戊二酰辅酶A脱氢酶(GCDH)缺乏导致戊二酸等有机酸在体内蓄积,引起脑白质及基底节损伤,其中苍白球受累较为常见。患儿多在出生后数月至1岁内起病,表现为巨颅、肌张力低下,随后出现肌张力增高、舞蹈样动作等锥体外系症状。早期诊断(通过新生儿筛查)及饮食控制(低赖氨酸、低色氨酸饮食)可显著改善预后,但若未及时治疗,苍白球损伤可导致严重的神经功能障碍。
3.丙酸血症与甲基丙二酸血症
均为常染色体隐性遗传的有机酸代谢障碍性疾病,因丙酸、甲基丙二酸等中间代谢产物蓄积,导致脑损伤。苍白球、丘脑等部位易受累,临床表现为呕吐、嗜睡、肌张力异常,严重者可出现昏迷及惊厥。急性期需纠正酸中毒、补充能量,长期需限制蛋白质摄入并补充特殊配方奶粉。
三、血管性与中毒性疾病:外源性因素导致的苍白球急性损伤
1.脑血管病
除前文所述的脑出血与脑梗死外,脑动静脉畸形(AVM)、烟雾病等脑血管异常也可累及苍白球。烟雾病患者因颈内动脉末端狭窄或闭塞,代偿性形成异常血管网,苍白球可因缺血或异常血管网出血而受损,临床表现为短暂性脑缺血发作(TIA)、脑卒中或运动障碍。
2.药物中毒
某些药物如抗精神病药物(如氟哌啶醇、氯丙嗪)长期大量使用,可导致药源性锥体外系反应,机制可能与阻断多巴胺受体、影响苍白球功能有关。患者表现为肌张力增高、震颤、静坐不能等,及时停药并给予抗胆碱能药物(如苯海拉明)可缓解症状。此外,海洛因、甲基苯丙胺等毒品滥用也可导致苍白球损伤,出现类似帕金森病的症状。
四、感染与免疫性疾病:炎症反应介导的苍白球损伤
1.中枢神经系统感染
细菌性脑膜炎:少数严重病例可并发基底节炎,苍白球受累时可出现肌张力异常及不自主运动。如肺炎链球菌脑膜炎患者,约5%会出现锥体外系症状,可能与炎症扩散及血管炎有关。
寄生虫感染:如脑囊虫病,囊虫可寄生在苍白球等部位,引起局部炎症反应及压迫症状,临床表现为癫痫发作、运动障碍。
2.自身免疫性疾病
系统性红斑狼疮(SLE):约10%-15%的SLE患者会出现神经精神症状,称为神经精神性狼疮(NPSLE),苍白球等基底节区可因免疫复合物沉积及小血管炎而受累,表现为舞蹈样动作、肌张力障碍。
抗磷脂抗体综合征(APS):可引起脑内小血管血栓形成,导致苍白球缺血性损伤,患者多表现为脑卒中、认知功能下降,部分可伴有运动障碍。
五、肿瘤与其他病变:占位效应或浸润导致的苍白球功能异常
1.原发性脑肿瘤
除胶质瘤、神经节细胞瘤外,苍白球还可发生其他类型肿瘤,如:
节细胞胶质瘤:由神经元和胶质细胞混合组成,生长缓慢,多表现为癫痫发作,少数可引起运动障碍。
淋巴瘤:原发性中枢神经系统淋巴瘤(PCNSL)可累及苍白球,患者多为免疫功能低下者,表现为迅速进展的认知障碍及神经功能缺损。
2.其他病变
钙化性病变:如Fahr病(特发性家族性脑血管亚铁钙沉着症),病因不明,可能与遗传或代谢异常有关,苍白球、丘脑等部位可出现对称性钙化,患者多在中年起病,表现为帕金森样症状、认知功能下降。
脱髓鞘疾病:如多发性硬化(MS),少数病例可累及苍白球,导致运动障碍,但相对少见,多以白质病变为主。
常见问题答疑
1.苍白球病变会遗传给下一代吗?
部分苍白球病变具有遗传性,如Wilson病、PKAN、亨廷顿病等,这些疾病多为常染色体显性或隐性遗传,有一定的遗传概率。以Wilson病为例,若父母双方均为携带者,子女患病概率为25%。而获得性苍白球病变(如缺血缺氧、中毒)通常不遗传。建议有家族史的人群进行基因检测,明确遗传风险,必要时进行遗传咨询及产前诊断。
2.如何早期发现儿童苍白球病变?
儿童苍白球病变的早期信号包括:运动发育迟缓(如4个月不能抬头、1岁不能独坐)、肌张力异常(肢体过硬或过软)、不自主运动(震颤、抽搐)、喂养困难及反复惊厥。家长若发现孩子出现上述症状,尤其是伴有家族史者,应及时带孩子到儿科神经内科就诊,进行头颅MRI、基因检测等检查,以便早期诊断。
3.苍白球病变患者的日常护理需要注意什么?
日常护理需根据患者具体情况制定:
运动障碍者:保持正确的体位,避免关节挛缩,定期进行被动运动及按摩;行走困难者需使用辅助器具(如轮椅、拐杖),预防跌倒。
吞咽困难者:给予软食或半流食,避免呛咳,必要时鼻饲喂养。
认知障碍者:进行认知训练(如记忆游戏、拼图),保持环境安静,减少刺激。
心理支持:关注患者情绪变化,鼓励参与社交活动,必要时进行心理疏导。
4.苍白球病变可以预防吗?
对于遗传性病变,目前尚无有效预防方法,但通过婚前检查、产前诊断可降低患病胎儿出生率;对于获得性病变,可通过以下措施预防:
围产期保健:避免宫内缺氧、产伤,降低新生儿HIE风险。
避免毒物接触:远离一氧化碳、锰等有毒物质,规范使用药物,避免滥用毒品。
控制基础疾病:如高血压、糖尿病患者需严格控制血压、血糖,降低脑血管病风险。
增强免疫力:预防感染,及时治疗病毒性脑炎、脑膜炎等疾病。
5.苍白球病变的治疗费用高吗?
治疗费用因病因、治疗方法而异:
遗传性疾病:如Wilson病,驱铜药物(青霉胺、曲恩汀)费用相对较低,每月约数百元,但需终身治疗;PKAN等罕见病尚无特效药物,对症治疗费用因方案不同而异。
获得性疾病:HIE的亚低温治疗费用约数千元,康复训练费用因疗程及机构不同,每月数千元至万元不等;DBS手术费用较高,单侧手术约10-15万元,双侧约20-30万元,但部分地区医保可报销。建议患者根据自身情况选择合适的治疗方案,并咨询当地医保政策。
- 文章标题:脑部苍白球的病变有哪些?小孩能改善吗?
- 更新时间:2025-05-23 15:25:38