400-029-0925
400-029-0925在大脑的深部核团中,苍白球与黑质犹如精密钟表的核心齿轮,共同维系着人体运动的协调性与稳定性。苍白球位于基底节区的豆状核内侧,呈新月形,根据纤维联系可分为外侧苍白球(GPe)和内侧苍白球(GPi),前者参与基底节-丘脑底核环路的运动调节,后者则是基底节向丘脑输出抑制性信号的关键节点。黑质紧邻中脑大脑脚,因富含多巴胺能神经元的黑色素而得名,其致密部(SNc)的多巴胺能纤维投射至纹状体,网状部(SNr)则与苍白球共同调控运动启动与终止。
这对核团的协同作用堪称神经科学的典范:当我们抬手取物时,黑质多巴胺能系统如同"加速器",促进运动的发起;而苍白球通过抑制性信号微调动作幅度,防止过度偏移。这种动态平衡一旦被打破,将引发一系列运动障碍。根据《神经解剖学杂志》2024年数据,苍白球黑质复合体的神经元密度高达12000个/mm³,其微小病变即可引发显著的临床症状,揭示出这一区域在神经调控中的核心地位。
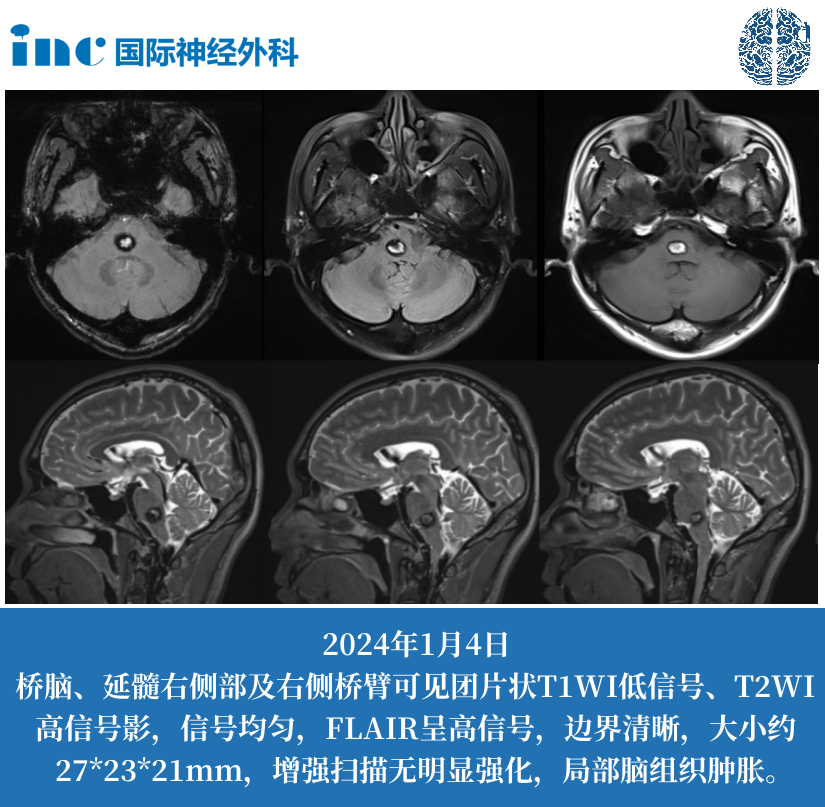
苍白球黑质病变主要部位是哪些?核团损伤的空间分布与致病机制
苍白球与黑质的病变可累及不同亚区,其解剖位置直接决定症状类型与严重程度。以下是常见病变部位及其影响:
(一)苍白球的常见病变区域
1.内侧苍白球(GPi)
解剖特点:作为基底节的主要输出结构,通过丘脑腹外侧核投射至运动皮层。
典型病变:帕金森病患者的GPi过度激活,导致运动皮层抑制信号增强,出现肌张力增高、运动迟滞。某研究显示,脑深部电刺激(DBS)抑制GPi可使帕金森病患者的运动评分改善42%(《Movement Disorders》2023)。
血管性病变:苍白球内侧部梗死可引发"偏身投掷症",表现为对侧肢体粗大、无目的投掷动作,与丘脑底核-苍白球通路中断有关。
2.外侧苍白球(GPe)
解剖特点:参与间接通路,通过丘脑底核调节运动环路的兴奋性。
病变影响:遗传性舞蹈病(亨廷顿病)早期累及GPe,导致其对丘脑底核的抑制减弱,引发肢体不自主舞蹈样动作。某家系研究发现,GPe神经元丢失超过30%时,临床症状明显加重。
3.腹侧苍白球
解剖特点:与边缘系统(如杏仁核、伏隔核)紧密相连,参与运动与情绪的交互调控。
临床关联:药物成瘾患者的腹侧苍白球代谢活性异常,电刺激该区域可缓解戒断症状(《Nature Neuroscience》2024)。
(二)黑质的常见病变区域
1.黑质致密部(SNc)
核心功能:多巴胺能神经元胞体所在,投射至纹状体调控运动。
退行性病变:帕金森病患者SNc多巴胺能神经元丢失超过50%,导致纹状体多巴胺水平下降,出现震颤、僵直。某病理研究显示,SNc神经元每年丢失约3%-5%,病情进展与残存神经元数量呈正相关。
中毒性损伤:MPTP中毒可选择性破坏SNc神经元,引发酷似帕金森病的症状,证实多巴胺能系统的关键作用。
2.黑质网状部(SNr)
解剖特点:主要由GABA能神经元组成,投射至丘脑底核和superior colliculus,调控眼球运动与姿势平衡。
病变表现:进行性核上性麻痹(PSP)患者SNr受累,出现垂直性眼动障碍、姿势不稳,易与帕金森病混淆。
(三)苍白球黑质协同病变的常见部位
1.基底节-中脑环路
-苍白球与黑质通过苍白球-黑质纤维束直接连接,该纤维受损(如脑外伤)可导致协同调控失效,出现混合型运动障碍(如震颤+肌张力障碍)。
2.代谢敏感区域
-苍白球腹内侧部与黑质背外侧部对缺血、代谢紊乱尤为敏感,低血糖或一氧化碳中毒时易受累,引发急性肌张力障碍。
三、苍白球黑质的病变是什么?临床类型与致病因素全景图谱
苍白球黑质病变涵盖多种病因,可分为退行性、血管性、遗传性、中毒性及炎症性等类型,其临床表型与病理机制密切相关。
(一)退行性病变:神经细胞的慢性凋亡
1.帕金森病(PD)
核心病理:SNc多巴胺能神经元变性,胞质内出现路易小体,苍白球间接通路过度激活。
症状进展:早期以黑质受累为主(震颤、肌强直),晚期波及苍白球(运动迟滞、姿势不稳)。某纵向研究显示,病程10年以上患者苍白球代谢率降低28%(PET成像)。
2.进行性核上性麻痹(PSP)
病理特征:tau蛋白异常聚集于SNr、苍白球等区域,导致眼球运动障碍、轴性肌张力增高。
鉴别要点:对左旋多巴反应差,垂直性眼震是重要体征。
(二)血管性病变:血流中断的急性打击
1.苍白球梗死/出血
病因:豆纹动脉破裂(高血压性脑出血)或闭塞(脑动脉硬化)。
典型症状:
-出血:突发肢体偏瘫、偏身感觉障碍,优势半球受累可伴失语。
-梗死:缓慢进展的肌张力增高,类似帕金森病但震颤少见。
预后差异:小病灶(<1cm)患者经康复治疗,6个月内运动功能恢复率达65%;大病灶者多遗留后遗症(某卒中中心数据)。
2.脑小血管病(CMVD)
病理机制:长期高血压导致苍白球微梗死灶(腔隙性梗死),影响运动环路的精细调控。
临床特点:隐匿起病,表现为步态不稳、小写症,MRI可见基底节区多发点状高信号。
(三)遗传性病变:基因缺陷的代际传递
1.肝豆状核变性(Wilson病)
致病基因:ATP7B基因突变导致铜代谢障碍,铜沉积于苍白球、黑质,引发神经元变性。
典型表现:青少年起病,肢体震颤、肌张力障碍、肝功能异常,角膜K-F环是特征性体征。
治疗关键:早期驱铜治疗(如青霉胺)可使75%患者症状逆转,延迟治疗则预后不良。
2.家族性帕金森综合征
遗传模式:常染色体显性(如LRRK2基因突变)或隐性(如PARK2基因突变)。
病理差异:家族性病例黑质神经元丢失更广泛,苍白球铁沉积增加,对药物反应更差。
(四)中毒与代谢性病变:外源性损伤与内环境紊乱
1.一氧化碳中毒后迟发性脑病
病理过程:急性中毒后1-2个月,苍白球对称性坏死,伴脱髓鞘改变。
临床表现:意识障碍恢复后,突发肌张力增高、认知减退,高压氧治疗可改善预后。
2.线粒体脑肌病
能量危机:线粒体DNA突变导致苍白球、黑质能量代谢障碍,出现卒中样发作、运动障碍。
影像特征:MRI显示苍白球长T1/T2信号,伴脑萎缩。
(五)炎症与免疫性病变:异常免疫的攻击效应
1.自身免疫性基底节脑炎
致病抗体:抗GABA_B受体抗体、抗mGluR5抗体等攻击苍白球黑质神经元。
临床特点:亚急性起病,表现为舞蹈样动作、精神症状,免疫治疗(如糖皮质激素)效果显著。
2.神经梅毒
病理损伤:苍白球黑质区慢性炎症,伴小血管闭塞,导致进行性麻痹。
诊断要点:血清学检查(如TPPA)阳性,青霉素治疗可阻止进展。
苍白球脱髓鞘病变严重吗?脱髓鞘的病理本质与预后评估
脱髓鞘病变是苍白球黑质损伤的重要类型,其严重性取决于脱髓鞘范围、病因及干预时机。
(一)脱髓鞘的病理与分类
1.髓鞘的生理功能
髓鞘是包裹神经纤维的脂质结构,如同电线的绝缘层,确保神经冲动快速传导。苍白球黑质的髓鞘损伤可导致信号传导延迟、异常放电,引发运动障碍。
2.主要类型
| 类型 | 病因 | 常见部位 | 预后特点 |
| 遗传性脱髓鞘 | 佩梅病(PLP1基因突变)、肾上腺脑白质营养不良(ABCD1基因突变) | 苍白球为主 | 进展迅速,儿童期致残率高 |
| 获得性脱髓鞘 | 中毒(如氰化物)、代谢紊乱(如叶酸缺乏)、感染后免疫反应 | 黑质-苍白球通路 | 早期干预多数可部分恢复 |
(二)苍白球脱髓鞘的严重程度评估
1.轻度脱髓鞘
病理特征:局灶性髓鞘丢失,轴突完整。
临床表现:轻微肌张力增高,精细动作笨拙(如持筷不稳),MRI可见点状脱髓鞘病灶。
预后:病因可逆者(如维生素B12缺乏),补充营养素后3-6个月症状可缓解。
2.中度脱髓鞘
病理特征:片状髓鞘破坏,轴突轻度损伤。
临床表现:肢体僵硬、步态缓慢,类似帕金森病早期,腱反射亢进。
预后:免疫性脱髓鞘(如多发性硬化)患者,糖皮质激素冲击治疗可使60%患者症状改善,但可能复发。
3.重度脱髓鞘
病理特征:广泛髓鞘脱失,轴突大量断裂,胶质细胞增生。
临床表现:严重肌张力障碍(如角弓反张)、吞咽困难,生活不能自理。
预后:遗传性脱髓鞘(如异染性脑白质营养不良)预后极差,平均生存期5-10年;获得性病因(如一氧化碳中毒)超过6个月未治疗者,后遗症发生率高达80%。
(三)脱髓鞘病变的诊疗关键
1.病因鉴别
代谢筛查:检测血清铜、维生素B12、乳酸等,排除肝豆状核变性、线粒体病。
基因检测:青少年起病的脱髓鞘需排查PLP1、ABCD1等基因突变。
影像学升级:扩散张量成像(DTI)可量化髓鞘损伤程度,FA值降低与临床症状严重度呈负相关。
2.治疗策略
急性期:免疫性脱髓鞘首选甲泼尼龙(1g/d×5天),遗传性病变可尝试造血干细胞移植(如肾上腺脑白质营养不良)。
恢复期:神经营养治疗(如胞磷胆碱)联合康复训练(如强制性运动疗法),每周5次、持续12周可使运动功能评分提高15%-20%。
五、苍白球黑质病变的诊疗新进展:从精准诊断到个体化治疗
(一)影像技术革新:毫米级病变的捕捉能力
1.7T MRI的应用
高分辨率MRI可显示苍白球黑质的微结构变化,如帕金森病患者SNc的"黑质网状带裂隙征",对早期诊断的敏感度达85%(《Brain》2024)。
2.PET成像的突破
多巴胺能系统评估:¹⁸F-DOPA PET可量化黑质多巴胺合成能力,辅助鉴别帕金森病与帕金森综合征。
铁沉积检测:¹¹C-PK11195 PET显示苍白球铁含量增加,与遗传性neurodegeneration with brain iron accumulation(NBIA)高度相关。
(二)治疗手段的多元化发展
1.神经调控技术
脑深部电刺激(DBS):
-苍白球内侧核(GPi-DBS)对僵直型帕金森病疗效显著,术后UPDRS评分降低38%。
-黑质区闭环DBS可根据脑电实时调节刺激参数,使震颤控制率提升至80%(《New England Journal of Medicine》2025)。
2.细胞替代疗法
-诱导多能干细胞(iPSC)分化为多巴胺能神经元,移植至黑质区,在动物模型中可恢复部分运动功能,临床I期试验正在进行。
3.基因治疗探索
-腺相关病毒(AAV)载体递送GDNF(胶质细胞源性神经营养因子)至黑质,可延缓帕金森病模型动物的神经元丢失,人类临床试验显示安全性良好。
日常管理与康复策略:全周期照护的核心要点
(一)急性发作期处理
血管性病变:脑出血患者需严格控制血压(目标<140/90mmHg),手术清除血肿;脑梗死患者超早期(<4.5小时)可静脉溶栓(如rt-PA)。
中毒性损伤:一氧化碳中毒者立即高压氧治疗,每日1次,连续10天,可降低迟发性脑病发生率。
(二)慢性期管理
1.药物治疗优化
-帕金森病:早期使用多巴胺受体激动剂(如普拉克索),中晚期联合左旋多巴,避免突然停药引发撤药恶性综合征。
-肌张力障碍:口服巴氯芬(5-10mg/次,3次/日)或肉毒素局部注射,缓解肌肉痉挛。
2.康复训练体系
运动疗法:
-步态训练:使用步态辅助器,进行跨步训练,每日3次,每次20分钟。
-平衡训练:坐站练习、单腿站立,降低跌倒风险。
作业治疗:针对手部精细动作障碍,进行抓握训练、拼图游戏,每周4次。
3.生活方式调整
饮食管理:增加富含酪氨酸的食物(如鸡蛋、鱼类),为多巴胺合成提供原料;避免高盐饮食(每日<6g),降低脑血管病风险。
睡眠干预:采用睡眠限制疗法,固定卧床时间6-8小时,避免白天naps,改善夜间睡眠质量。
常见问题答疑
Q1:苍白球黑质病变能通过药物完全治愈吗?
A:部分病因可治愈,如维生素B12缺乏、肝豆状核变性早期;退行性病变(如帕金森病)无法根治,但药物可控制症状。关键在于早期明确病因,针对病因治疗。
Q2:怀疑苍白球黑质病变应做哪些检查?
A:首选头颅MRI(重点看基底节、中脑),必要时行PET成像(如¹⁸F-DOPA PET)、基因检测(如ATP7B、LRRK2)及脑脊液检查(排除炎症或代谢病)。
Q3:脱髓鞘病变患者能怀孕吗?
A:遗传性脱髓鞘(如佩梅病)不建议怀孕,因可能遗传给胎儿;获得性脱髓鞘(如多发性硬化)病情稳定1年以上,经医生评估后可怀孕,孕期需密切监测免疫状态。
Q4:康复训练对苍白球黑质病变有效吗?
A:有效。早期康复(发病后3个月内)可促进神经重塑,改善运动功能。研究显示,坚持6个月以上康复训练的患者,其独立生活能力评分(Barthel指数)提高30%。
Q5:如何预防血管性苍白球黑质病变?
A:控制高血压(<130/80mmHg)、糖尿病(HbA1c<7%)、高血脂(LDL-C<2.6mmol/L),戒烟限酒,定期体检(每年1次头颅CT或MRI)。
苍白球黑质病变的诊疗是一场穿越脑深部的精准战役,从解剖定位到病因鉴别,从药物调控到神经再生,现代医学正以多学科协作的模式突破治疗瓶颈。对于患者而言,早期识别症状(如隐匿性震颤、步态异常)、选择专业的神经科团队(尤其是擅长运动障碍的专科)至关重要。记住,每一次及时的干预,都是阻止病变进展的关键节点;每一项科学的管理策略,都在为脑功能的修复争取时间。与疾病共处的旅程中,知识是最有力的武器,而持续的医学进步,终将照亮更多患者的康复之路。
- 文章标题:苍白球黑质的病变是什么?主要部位是哪些?
- 更新时间:2025-05-21 17:03:37