400-029-0925
400-029-0925众所周知,胶质瘤是较常见的颅内肿瘤,然而有一大类罕神经元-胶质肿瘤却是少见而复杂多样的,多数作者认为这是一种有神经胶质细胞与神经元瘤细胞的良性混合性肿瘤。神经元肿瘤和混合性神经元-胶质肿瘤神经元肿瘤和混合性神经元-胶质肿瘤的特征是不同程度的神经元和胶质分化。这类肿瘤包括:胚胎发育不良性神经上皮肿瘤(dysembryoplastic neuroepithelial tumor, DNT/DNET)、节细胞胶质瘤和节细胞瘤、小脑发育不良性节细胞瘤(Lhermitte-Duclos病)、婴幼儿促纤维增生性星形细胞瘤/节细胞胶质瘤、乳头状胶质神经元肿瘤、菊形团形成性胶质神经元肿瘤、弥漫性柔脑膜胶质神经元肿瘤、小脑脂肪神经细胞瘤、中央神经细胞瘤和脑室外神经细胞瘤,以及副神经节瘤。
胚胎发育不良性神经上皮肿瘤 — DNT
DNT系生长缓慢的幕上肿块,通常发生在伴长期难治性局灶性癫痫史的儿童和年轻成人中。除了癫痫发作,DNT患者通常几乎没有神经系统体征,智力通常正常。透明隔中的少见DNT可能会引起脑积水和颅内压增高。
DNT的关键病理特征包括:存在被肿瘤细胞包围的皮质神经元、发育不良的皮质结构病灶、成分类似于星形细胞瘤或少突神经胶质瘤的多结节结构以及与皮质表面垂直的柱状结构。需将DNT与弥漫性胶质瘤相鉴别。约30%的DNT存在BRAF V600E突变。透明隔DNT常存在血小板衍生生长因子受体A(PDGFRA)突变。也可见成纤维细胞生长因子受体1(FGFR1)的基因改变。 IDH1型和2型突变都不符合DNT,存在任一突变都提示弥漫性胶质瘤,同样1p和19q共缺失也提示弥漫性胶质瘤。
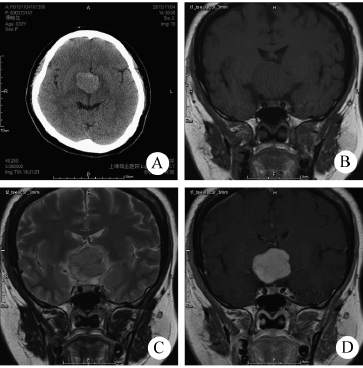
DNT在MRI T1加权像上表现为低信号,在MRI T2加权像上表现为高信号。病灶基于皮质,并局部扩张皮质,有时会延伸入白质。增强表现各异,发生于不到1/2的病例;增强为斑片状、多灶性,而非弥漫性增强。因肿瘤生长缓慢,邻近病灶的颅骨有时变形。
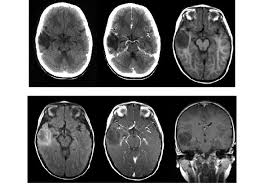
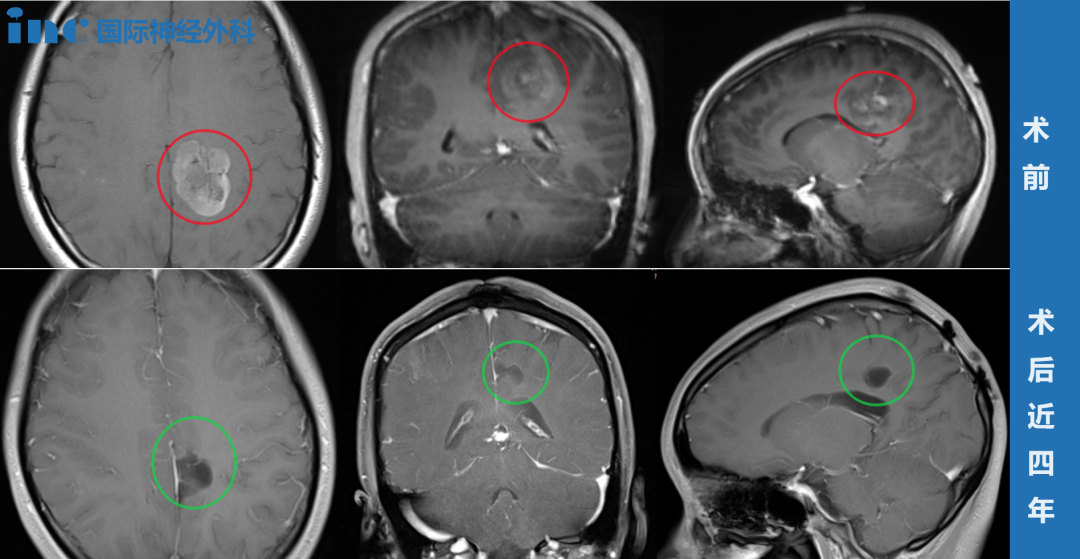
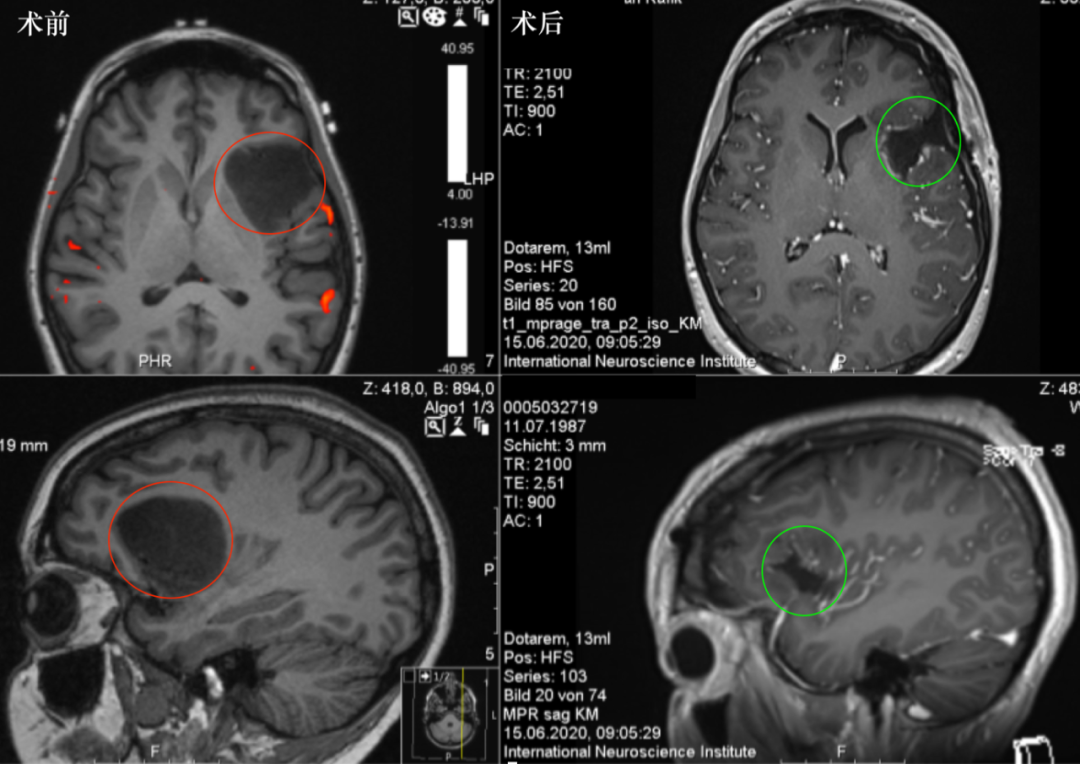
图:DNET磁共振表现
手术指征是难治性癫痫发作,但如果可能,对皮层功能区的病变较好进行观察。大多数患者至少在手术后初期无癫痫发作。然而,随着随访持续时间延长,癫痫发作的发生率似乎会增加。一项研究阐明了这一点,该研究纳入26例儿童,其中62%的儿童在中位随访4.3年时仍无癫痫发作]。癫痫发作复发的主要危险因素为年龄>10岁和术前癫痫史较长(>2年)。少见情况下可能出现肿瘤复发,甚至恶变。
节细胞胶质瘤和节细胞瘤
— 节细胞胶质瘤和节细胞瘤包括一系列以发育不良性神经元群为特征的低级别肿瘤。在节细胞胶质瘤中,发育不良的神经元细胞伴随着肿瘤性胶质细胞,而在节细胞瘤中,分化良好的较大神经元是的肿瘤成分。
尽管这些肿瘤可能出现在整个中枢神经系统的任何地方,但是大多数是幕上肿瘤且位于颞叶。高达60%的节细胞胶质瘤存在BRAF V600E突变,含此突变的儿科低级别胶质瘤经标准治疗后复发风险较高。突变的蛋白主要见于神经元细胞中。低级别中线节细胞胶质瘤的胶质成分和神经元成分可能都存在H3F3A K27M突变和BRAF V600E突变。除了BRAF突变,发现高达30%的胶质神经元肿瘤存在神经营养性受体酪氨酸激酶(neurotrophic receptor tyrosine kinase, NTRK)等基因的致瘤融合,该突变可能具有治疗意义。
若在组织学确定为节细胞胶质瘤的肿瘤中发现IDH1突变,则强烈支持弥漫性胶质瘤的诊断,而且患者往往诊断时年龄较大,复发风险较高,预后较差。节细胞胶质瘤和节细胞瘤通常出现在儿童和年轻成人中;大部分病例系列研究中患者的平均年龄约为20岁。癫痫发作是较常见的主诉症状,并且频发长时间癫痫。节细胞胶质瘤可为行颞叶切除术治疗癫痫时的偶然发现。
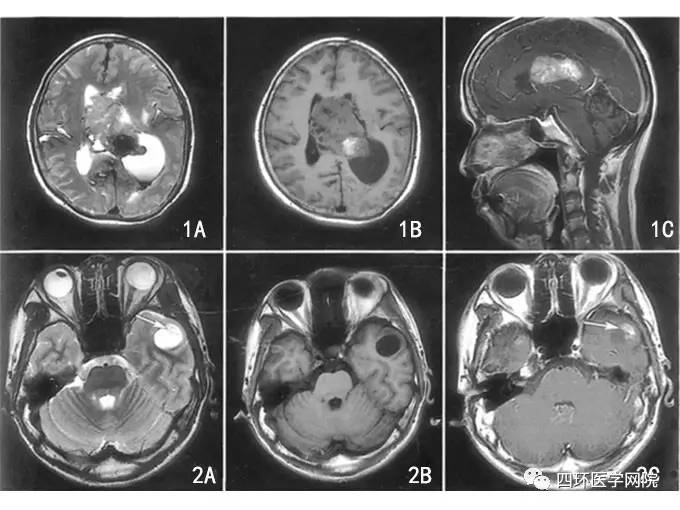
这些肿瘤在MRI T2加权像上表现为高信号。1/2的病例存在囊肿,约1/3的病例存在钙化。对比增强程度不一,可能无强化,也可能明显强化,可能是病灶实体或边缘强化。
初始治疗是外科切除。虽然肉眼下全切较理想,但即使是次全切,预后也较好。两种情况下都可出现远期复发。一项病例系列研究阐明了这一点,该研究纳入62例低级别节细胞胶质瘤成人患者,其中肉眼下全切后和次全切后患者的5年无进展生存率分别是78%和62%。肿瘤出现进展的时间为2个月到20年,对于诊断时年龄超过40岁的患者,该时间更短(14年 vs 3年)。
虽然不到10%的节细胞胶质瘤会变为间变性肿瘤,但节细胞胶质瘤出现间变是一种恶兆。即使进行的放疗和化疗,间变性节细胞胶质瘤也通常致命。越来越多的证据表明,BRAF控制剂单药治疗或与丝裂原活化蛋白激酶激酶(mitogen-activated protein kinase kinase, MEK)控制剂联用,对存在BRAF V600E突变的肿瘤有临床效果。此外,还有动物模型证据表明BRAF突变本身会导致这些肿瘤患者出现癫痫;因此,BRAF控制剂对难治性癫痫可能有益。NTRK控制剂恩曲替尼(entrectinib)和拉罗替尼(larotrectinib)获美国FDA批准用于既往标准治疗无效或不可切除的NTRK融合阳性肿瘤患者。
对于已接受完全切除术的患者,不放疗。尽管人们担心放疗可能促使其转化为间变程度更高的肿瘤,但一项回顾性文献综述提示,次全切后的放疗好转了低级别和高级别节细胞胶质瘤的局部肿瘤控制。然而,未显示放疗对总体生存率有影响。关于低级别节细胞胶质瘤次全切后放疗的作用,这些数据不足以给出强有力的。
小脑发育不良性节细胞瘤
— 小脑发育不良性节细胞瘤(也称Lhermitte-Duclos病)是一种较其少见的疾病,以正常小脑皮质结构丧失和小脑小叶局灶性增厚为特征。目前尚不清楚小脑发育不良性节细胞瘤是小脑皮质的瘤性还是错构性肿瘤;如果是瘤性,则对应WHO分类Ⅰ级;其组织学表现为小脑白质减少,并存在异常肥大的神经节细胞,与浦肯野细胞颇为相似。
小脑发育不良性节细胞瘤通常见于年轻成人和中年人,但在年龄低至3岁和高至70岁的患者中也被诊断出。小脑症状可能在确诊前已存在数年,常有梗阻性脑积水。病变出现在小脑半球,偶尔延伸入小脑蚓。该病伴发育异常(包括大头畸形和精神发育迟滞)的情况并不少见。
小脑发育不良性节细胞瘤可呈家族性或散发性方式发病。针对小脑发育不良性节细胞瘤患者的分子研究提示,PTEN/AKT通路(细胞生长的主要调节器)出现异常的频率较高。同样,该肿瘤与Cowden综合征有关,这种常染色体显性遗传病的特征是多发性错构生长病灶,乳腺癌、子宫癌和甲状腺癌的发病率增高,以及PTEN基因种系突变。
大多数有发育不良性节细胞瘤的成人(而非儿童)具有PTEN种系突变。俄亥俄州立大学修订了临床诊断标准,将小脑发育不良性节细胞瘤作为Cowden综合征的一个主要标准,而美国国家综合癌症网络将其作为基因检测Cowden综合征的一个指征,Cowden综合征是一种常染色体显性疾病,具有年龄相关的外显率和不同的表现方式。Cowden综合征的易感基因PTEN位于染色体10q23.3。
小脑发育不良性节细胞瘤的MRI特征是小脑小叶扩大伴结构扭曲,以及不同程度的囊性变。增强MRI扫描病灶通常无强化,T1像呈低信号,T2像以高信号与低信号交替的层状模式为特征。肿块为局限性,与周围组织界限分明。在少数病例中,对比增强扫描出现强化,这可能代表小脑皮质的外层有静脉增生和的引流静脉。
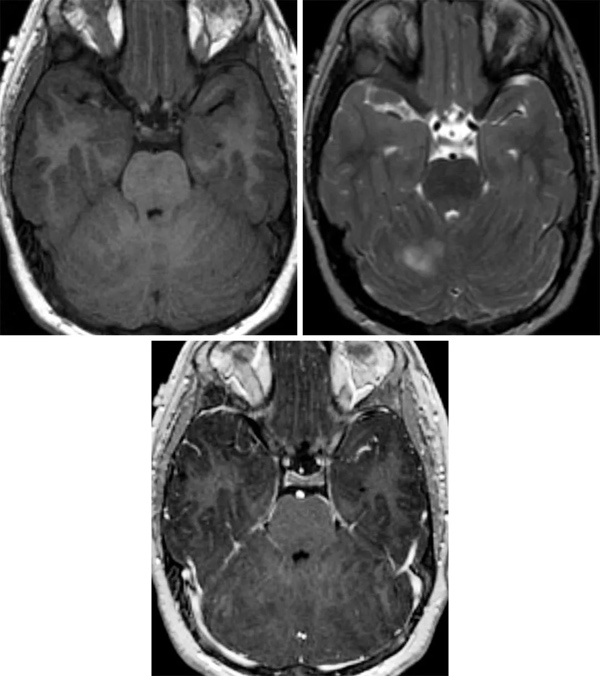
小脑发育不良性节细胞瘤的治疗方法为外科切除,不过,少数病例在明显肉眼下全切后复发。采用雷帕霉素控制PI3K/PTEN/AKT通路对1例有严重症状性双侧发育不良性节细胞瘤的婴儿合适。由于该病在成人中与Cowden综合征有关,医生应排除合并恶性肿瘤(是乳腺癌和泌尿生殖系癌症),并安排患者行Cowden综合征的基因检测。
婴幼儿促纤维增生性星形细胞瘤/节细胞胶质瘤 — 婴幼儿促纤维增生性星形细胞瘤和婴幼儿促纤维增生性节细胞胶质瘤最初被认为只发生于2岁以内,但后续有25岁患者发生该病的报道。该病的典型特征包括:星形细胞与神经节双重分化、的促纤维增生基质、瘤体大、位于大脑半球、存在实性和囊性成分、在T2加权MRI上明显的低信号,预后良好。已有存在BRAF V600E突变少见病例的报道。治疗方法为手术,在不完全切除的情况下应联合化疗。大多数研究表明,肉眼下全切可带来长期存活。通常只有在已用尽其他方法后才考虑放疗。
乳头状胶质神经元肿瘤
乳头状胶质神经元肿瘤是少见的低级别肿瘤,主要发生于年轻成人,诊断时中位年龄为23岁。这些肿瘤的特征性表现包括:位于大脑半球、包含星形细胞和神经元分化,以及独特的假乳头状结构模式。这类肿瘤患者发生SLC44A1-PRKC融合并引起丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)信号失调;虽然这种融合较少见,但可能是疾病的特异性特征。
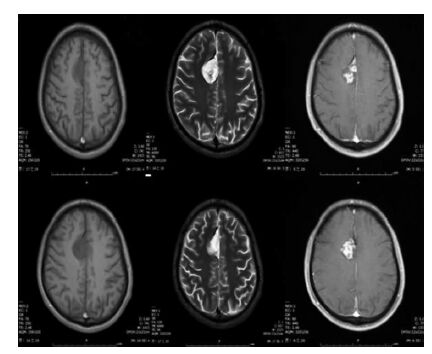
MRI特征与其他神经元-胶质肿瘤类似。大多数病变有囊性成分和可强化的实性成分,其T2加权像表现为等信号高信号。也可能观察到钙化。在大多数病例中,乳头状胶质神经元肿瘤可通过外科切除治愈。少见的复发性或进行性乳头状胶质神经元肿瘤也有报道,通常与Ki-67指数升高有关(>5%)。
菊形团形成性胶质神经元肿瘤
— 菊形团形成性胶质神经元肿瘤是一种生长缓慢的少见病变,主要见于年轻成人。这类肿瘤通常发生在四脑室,但也可见于幕上脑室系统、小脑、松果体区、视交叉、脊髓、透明隔和脑实质。
典型组织病理学特征是神经细胞(排列成假菊形团)和毛细胞型星形胶质细胞的混合群。不同于毛细胞型星形细胞瘤,菊形团形成性胶质神经元肿瘤不存在BRAF基因改变;部分病例存在PIK3CA或FGFR1突变。
发生在四脑室的肿瘤通常表现为脑积水,因此常见头痛。在MRI扫描上,表现为不均匀、囊性或多房性病灶,伴强化区域。在一些病例中,侵犯小脑或脑干的实质。大多数病例似乎可通过手术切除治愈,但也有少见病例出现肿瘤播散至整个脑室系统的报道。
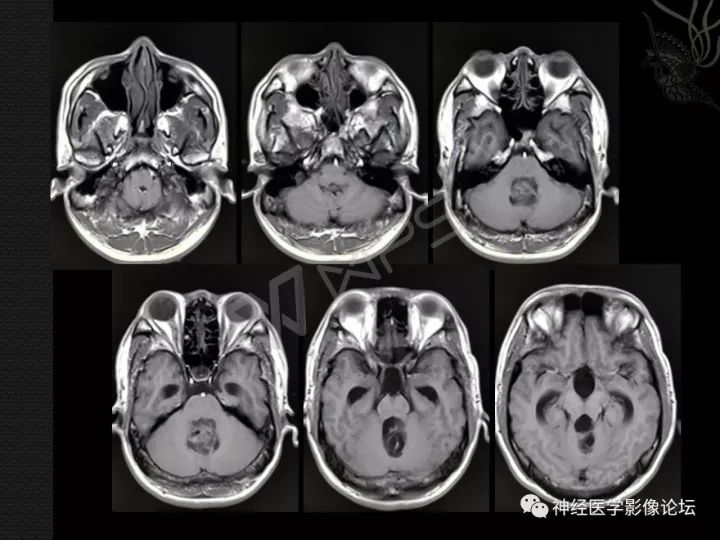
弥漫性柔脑膜胶质神经元肿瘤
弥漫性柔脑膜胶质神经元肿瘤是WHO于2016年对CNS肿瘤分类系统修订后新指定的肿瘤。过去的报告用其他名称描述该肿瘤,包括原发性柔脑膜少突神经胶质瘤病和播散性少突神经胶质瘤样柔脑膜肿瘤。典型特征为且广泛的柔脑膜生长、少突胶质细胞样细胞学,以及部分病例有神经元分化证据。这些肿瘤的基因特征通常包括合并BRAF-KIAA1549基因融合及1p缺失或1p/19q共缺失,不存在IDH突变。较少报道的异常有H3K27M和FGFR1突变。
根据已发表的较大型研究(36例),诊断时的中位年龄为5岁(5月龄-45岁)。大多数患者会急性出现脑积水致颅内压增高的症状和体征、颅神经病变和其他柔脑膜体征。
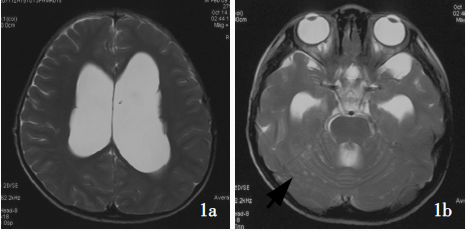
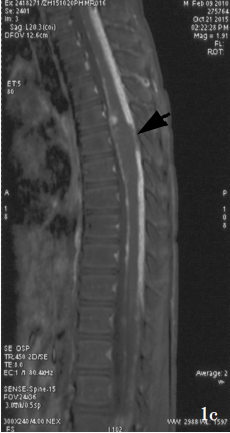
MRI显示广泛的柔脑膜强化和增厚,通常沿脊髓、颅后窝和脑干较为明显,也可见实质内和髓内的强化结节。虽然蛋白水平增高,但脑脊液细胞学检查通常呈阴性,诊断通常需要脑膜活检。该肿瘤细胞的组织病理学常表现为低级别特征,但尚未确定其WHO分级。
最佳治疗方法尚不清楚,这些肿瘤的病程相对惰性。由于很多患者在诊断时的年龄较小,通常在放疗之前先尝试化疗。一项研究纳入了36例患者并中位随访5年,中位生存期尚未达到;在9例死亡的患者中,一次活检后的生存期为<1年到21年不等(中位3年)。核分裂活性、Ki67>4%和微血管增生与生存较差有关。
中央神经细胞瘤和脑室外神经细胞瘤
— 中央神经细胞瘤是分化良好的肿瘤,约占成人全部脑室内肿瘤的一半。在脑实质或脊髓中偶尔可发现类似的肿瘤,在这种情况下被称为脑室外神经细胞瘤。虽然其通常位于脑室内,但是少侵袭柔脑膜。
中央神经细胞瘤是神经外胚叶肿瘤,有的神经元分化。特征为突触素染色呈弥漫性阳性,并且大多对NeuN具有免疫反应性。对20例典型的中央神经细胞瘤患者进行遗传研究,识别出MYCN、PTEN和OR5BF1的过表达以及BIN1、SNRPN和HRAS的表达不足,这些结果提示,MYCN的过表达和抑癌基因BIN1的表达减少可能促发肿瘤。一项纳入7例脑室外神经细胞瘤的研究发现,不存在IDH1/IDH2突变和MGMT启动子甲基化。
中央神经细胞瘤患者的平均诊断年龄是29岁。大多数患者表现出脑积水所致的颅内压增高的症状。也可能出现视觉障碍和认知功能障碍。局灶性神经功能障碍并不常见。少数患者出现脑室内出血。
大多数中央神经细胞瘤是多囊性肿瘤且有钙化,以宽基底附着在脑室上外侧壁上。它们通常位于侧脑室或三脑室,附着于室间孔附近的透明隔或脑室壁上。但通常不会出现在侧脑室枕角或颞角。
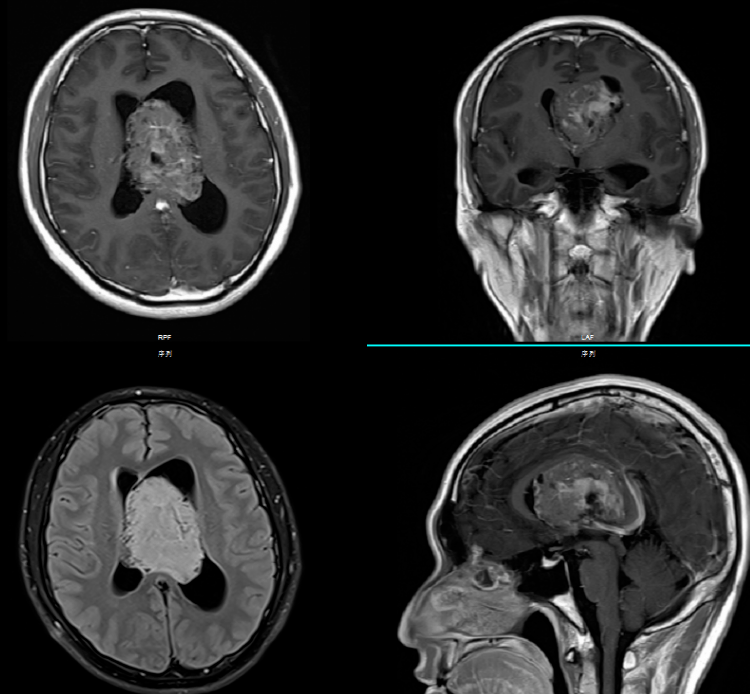
典型的CT表现是脑室内不均匀的高密度肿块,伴有中等对比增强。MRI T1加权像表现为轻度高信号,T2加权像表现在某种程度上更加多变。使用钆后通常增强。可能难以通过MRI区分中央神经细胞瘤、脑室外神经细胞瘤与更常见的病灶(像高级别胶质瘤)。
最佳治疗方法是完全手术切除。由于这些肿瘤通常再生长缓慢,即使是次全切除也可能延长生存期。典型神经细胞瘤(即MIB-1单克隆抗体检测到生长分数<3%)患者似乎预后,辅助放疗或立体定向放射外科对不完全切除的患者或具有非典型组织学的患者可能有用。对肿瘤复发或进展通常采用局部放疗。至少一项报道提示,复发的中央神经细胞瘤可能对全身化疗有反应。
一项回顾性病例系列研究纳入在35年间使用手术、放疗和/或化疗进行治疗的45例患者,该研究中的治疗结果阐明了中央神经细胞瘤治疗的结局和预后因素。相比病变不典型的患者,典型神经细胞瘤患者的10年生存率(90% vs 63%)和局部控制率(74% vs 46%)更好。术后放疗相比未行术后放疗,术后放疗能够提高患者10年时的局部控制率(75% vs 51%),但这未能转化为总体生存获益。一项纳入71例患者的多中心研究对肿瘤进行了免疫组化评估,结果发现手术程度是的预后指标。然而,该研究的病例数量太少可能不足以确定MIB-1指数的预后阈值。
小脑脂肪神经细胞瘤
小脑脂肪神经细胞瘤是颅后窝肿瘤,患者在诊断时的平均年龄约为50岁。肿瘤包含成熟脂肪组织和多种其他细胞类型,包括程度的神经元分化。基因检测已证明,小脑脂肪神经细胞瘤为一种独特的临床疾病类型,其与神经细胞瘤的关系比与髓母细胞瘤的关系更密切。脂肪酸结合蛋白4(FAB4)的过表达可能帮助区分这些肿瘤与髓母细胞瘤。
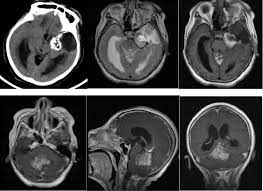
CT表现为界限清楚的病灶(低密度或等密度影),并且有中度不均匀强化。MRI显示肿瘤为T1低信号,T2不均匀高信号,而使用钆后表现为不均匀强化。已报道的病例大约只有40例,因此最佳治疗和总体预后难以估计。小脑脂肪神经细胞瘤的自然病程似乎相对较长。鉴于此特点,手术治疗,如果疾病复发,可再次行手术或放疗。然而,在一些病例中观察到更具侵袭性的自然病程。
颈静脉鼓室副神经节瘤
— 颈静脉鼓室副神经节瘤(以前称为颈静脉球瘤或鼓室球瘤)富含血管,源于副神经节组织(通常是副交感神经起源),通常为良性肿瘤。约1/3的病例出现在遗传综合征中,可进行相应的基因检测。在CNS中,副神经节瘤也可能出现在马尾/终丝。
室管膜下巨细胞星形细胞瘤
— SEGA是生长缓慢的良性胶质肿瘤,见于结节性硬化症患者。SEGA通常起于脑室周围区域。虽然这些肿瘤已被归入星形细胞瘤,但是它们由胶质神经元系混合而成,更准确的称谓是室管膜下巨细胞肿瘤(subependymal giant cell tumor, SGCT)。雷帕霉素机能靶点(mechanistic target of rapamycin, mTOR)控制剂(如西罗莫司和依维莫司)对这些肿瘤可能合适。
星形母细胞瘤
— 星形母细胞瘤是少见的胶质肿瘤,具有星形细胞、室管膜和其他神经胶质的重叠成分。星形母细胞瘤通常见于年龄较大的儿童和年轻成人,不过也有71岁患者的报道。
其MRI表现为分散的、分叶状、幕上病灶,含实性和囊性成分。实性成分已被描述为在T2加权序列上具有特征性“多泡”外观。常常记录到T2低信号,对比强化和钙化情况多变。
病理学上,星形母细胞瘤兼有星形细胞瘤和室管膜瘤的特征;但不同于室管膜瘤,大多星形母细胞瘤Olig2染色呈阳性。特征性的表现是弥散性的血管周围星形母细胞性假菊形团。存在低级别和高级别变异型,且分级和核分裂指数似乎与预后相关。与其他主要基于皮质、界限清楚的胶质瘤一样,约1/3的星形母细胞瘤存在BRAF V600E突变,但不存在IDH1 R132H突变。
低级别病灶的肉眼下全切通常带来长时间的无病间期。高级别病灶的治疗方法包括手术、放疗和化疗,不过辅助治疗的效果存在争议。与年轻患者相比,30岁以后确诊患者的总生存情况似乎更差。
三脑室脊索样胶质瘤
— 三脑室脊索样胶质瘤见于成人,平均年龄46岁,男女比例为1:2。因其位于三脑室,患者常出现脑积水的症状。
在MRI上,这些肿瘤与脑实质界限分明,但可能侵犯下丘脑。病灶在T1加权序列上是等信号,且通常为致密均匀的增强表现。
在病理学检查上,存在神经胶质抗原染色的索状和簇状上皮样细胞。尚未发现特征性遗传改变。不存在高级别肿瘤特征。肉眼下全切可治愈该病,分割放疗或立体定向放射外科对于不完全切除的病灶可能具有作用。肿瘤及其治疗的并发症可能包括尿崩症和记忆障碍。
血管中心性胶质瘤
— 血管中心性胶质瘤主要见于儿童和年轻成人,诊断时平均年龄为17岁。大多数病例表现出难治性癫痫发作。肿瘤位于大脑半球表面。
越来越多的证据指出,MYB基因家族的改变是血管中心性胶质瘤分子发病机制的重要。一项研究显示,19例血管中心性胶质瘤均有MYB基因改变,其中7例进行了全基因组或RNA测序,发现6例存在MYB基因与QKI基因(一种抑癌基因)的重现性染色体内融合。
其MRI扫描表现为分散的非强化病灶,在T2加权序列上呈高信号。在一些病例中观察到独特的MRI特征,包括T1加权序列上皮质脑回边缘的固有高信号和从肿瘤延伸到脑室壁的T2高信号带。单形双较的肿瘤细胞在胶质抗原和室管膜抗原染色中表达程度各异,并显示出血管中心性生长模式。
手术通常可治愈该病。
总结
●脑肿瘤是一组多种多样的肿瘤,源于中枢神经系统(CNS)内不同细胞,或源于转移到CNS的全身性肿瘤。较常见的脑肿瘤包括弥漫性胶质瘤、脑膜瘤和来自全身肿瘤的转移瘤。
●一般而言,少见脑肿瘤的临床表现取决于解剖部位、生长速度和组织学。尽管患者的症状、影像学特征、人口统计学特征可能提示为特定的瘤种,但脑肿瘤的明确诊断需进行活检。
●神经元肿瘤和混合性神经元-胶质肿瘤的特征是不同程度的神经元和胶质分化。多数为低级别、界限清楚、基于皮质的肿瘤,伴有癫痫发作。BRAF基因改变和致癌融合事件在某些亚型中很常见,包括节细胞胶质瘤和胚胎发育不良性神经上皮肿瘤(DNT)。
●对于大多数少见脑肿瘤,尽量优选手术切除整个肿瘤。视具体情况选择辅助疗法(放疗和/或化疗),通常仅用于自然病程具有侵袭性且预后较差的肿瘤。基因测序的应用增加,可能会发现特定瘤种的基因改变,有助于选择靶向疗法,并获得更好的结局
参考来源:RARE BRAIN TUMORS.WWW.UPTODATE.COM
- 文章标题:那些少见的脑瘤|神经元-胶质肿瘤咨询解读
- 更新时间:2021-04-14 10:36:30